
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Учебное пособие: Коллоидная химия
Учебное пособие: Коллоидная химия
ВВЕДЕНИЕ
Предметом физической химии является объяснение химических явлений на основе более общих законов физики. Физическая химия рассматривает две основные группы вопросов:
1. Изучение строения и свойств вещества и составляющих его частиц;
2. Изучение процессов взаимодействия веществ.
В курсе физической химии обычно выделяют несколько разделов.
Строение вещества. В этот раздел входят учение о строении атомов и молекул и учение об агрегатных состояниях вещества. Изучение строение вещества необходимо для выяснения важнейших вопросов об образовании молекул из атомов, о природе химической связи, о строении и взаимодействии молекул. Именно в этой своей части физическая химия очень тесно переплетается со всеми направлениями современной химии, поскольку изучение химических свойств вещества вне связи со строением атомов и молекул на современном уровне невозможно.
Химическая термодинамика изучает энергетические эффекты химических процессов; позволяет определить возможность, направление и глубину протекания химического процесса в конкретных условиях.
Химическая кинетика. В этом разделе физической химии изучается скорость и механизм протекания химических процессов в различных средах при различных условиях.
Учение о растворах рассматривает процессы образования растворов, их внутреннюю структуру и важнейшие свойства, зависимость структуры и свойств от природы компонентов раствора.
Электрохимия изучает особенности свойств растворов электролитов, явления электропроводности, электролиза, коррозии, работу гальванических элементов.
Коллоидная химия изучает поверхностные явления и свойства мелкодисперсных гетерогенных систем.
Все разделы физической химии объединяет единая основа – общие законы природы, которые применимы к любым процессам и любым системам, независимо от их строения.
1 ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Термодинамика – наука о взаимопревращениях различных форм энергии и законах этих превращений. Термодинамика базируется только на экспериментально обнаруженных объективных закономерностях, выраженных в двух основных началах термодинамики.
Термодинамика изучает:
1. Переходы энергии из одной формы в другую, от одной части системы к другой;
2. Энергетические эффекты, сопровождающие различные физические и химические процессы и зависимость их от условий протекания данных процессов;
3. Возможность, направление и пределы самопроизвольного протекания процессов в рассматриваемых условиях.
Необходимо отметить, что классическая термодинамика имеет следующие ограничения:
1. Термодинамика не рассматривает внутреннее строение тел и механизм протекающих в них процессов;
2. Классическая термодинамика изучает только макроскопические системы;
3. В термодинамике отсутствует понятие "время".
1.1 ОСНОВНЫЕ ПОНЯТИЯ ТЕРМОДИНАМИКИ
Термодинамическая система тело или группа тел, находящихся во взаимодействии, мысленно или реально обособленные от окружающей среды.
Гомогенная система – система, внутри которой нет поверхностей, разделяющих отличающиеся по свойствам части системы (фазы).
Гетерогенная система система, внутри которой присутствуют поверхности, разделяющие отличающиеся по свойствам части системы.
Фаза – совокупность гомогенных частей гетерогенной системы, одинаковых по физическим и химическим свойствам, отделённая от других частей системы видимыми поверхностями раздела.
Изолированная система система, которая не обменивается с окружающей средой ни веществом, ни энергией.
Закрытая система – система, которая обменивается с окружающей средой энергией, но не обменивается веществом.
Открытая система – система, которая обменивается с окружающей средой и веществом, и энергией.
Совокупность всех физических и химических свойств системы характеризует её термодинамическое состояние. Все величины, характеризующие какое-либо макроскопическое свойство рассматриваемой системы – параметры состояния. Опытным путем установлено, что для однозначной характеристики данной системы необходимо использовать некоторое число параметров, называемых независимыми; все остальные параметры рассматриваются как функции независимых параметров. В качестве независимых параметров состояния обычно выбирают параметры, поддающиеся непосредственному измерению, например температуру, давление, концентрацию и т.д. Всякое изменение термодинамического состояния системы (изменения хотя бы одного параметра состояния) есть термодинамический процесс. Обратимый процесс – процесс, допускающий возможность возвращения системы в исходное состояние без того, чтобы в окружающей среде остались какие-либо изменения.
Равновесный процесс процесс, при котором система проходит через непрерывный ряд равновесных состояний.
Энергия – мера способности системы совершать работу; общая качественная мера движения и взаимодействия материи. Энергия является неотъемлемым свойством материи. Различают потенциальную энергию, обусловленную положением тела в поле некоторых сил, и кинетическую энергию, обусловленную изменением положения тела в пространстве.
Внутренняя энергия системы сумма кинетической и потенциальной энергии всех частиц, составляющих систему. Можно также определить внутреннюю энергию системы как её полную энергию за вычетом кинетической и потенциальной энергии системы как целого.
Формы перехода энергии от одной системы к другой могут быть разбиты на две группы. В первую группу входит только одна форма перехода движения путем хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота. Теплота есть форма передачи энергии путём неупорядоченного движения молекул. Во вторую группу включаются различные формы перехода движения, общей чертой которых является перемещение масс, охватывающих очень большие числа молекул (т.е. макроскопических масс), под действием каких-либо сил. Таковы поднятие тел в поле тяготения, переход некоторого количества электричества от большего электростатического потенциала к меньшему, расширение газа, находящегося под давлением и др. Общей мерой передаваемого такими способами движения является работа форма передачи энергии путём упорядоченного движения частиц.
Теплота и работа характеризуют качественно и количественно две различные формы передачи движения от данной части материального мира к другой. Теплота и работа не могут содержаться в теле. Теплота и работа возникают только тогда, когда возникает процесс, и характеризуют только процесс. В статических условиях теплота и работа не существуют. Различие между теплотой и работой, принимаемое термодинамикой как исходное положение, и противопоставление теплоты работе имеет смысл только для тел, состоящих из множества молекул, т.к. для одной молекулы или для совокупности немногих молекул понятия теплоты и работы теряют смысл. Поэтому термодинамика рассматривает лишь тела, состоящие из большого числа молекул, т.е. так называемые макроскопические системы.
1.2 ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Первое начало термодинамики представляет собой закон сохранения энергии, один из всеобщих законов природы (наряду с законами сохранения импульса, заряда и симметрии):
Энергия неуничтожаема и несотворяема; она может только переходить из одной формы в другую в эквивалентных соотношениях.
Первое начало термодинамики представляет собой постулат – оно не может быть доказано логическим путем или выведено из каких-либо более общих положений. Истинность этого постулата подтверждается тем, что ни одно из его следствий не находится в противоречии с опытом. Приведем еще некоторые формулировки первого начала термодинамики:
Полная энергия изолированной системы постоянна;
Невозможен вечный двигатель первого рода (двигатель, совершающий работу без затраты энергии).
Первое начало термодинамики устанавливает соотношение между теплотой Q, работой А и изменением внутренней энергии системы ΔU:
Изменение внутренней энергии системы равно количеству сообщенной системе теплоты минус количество работы, совершенной системой против внешних сил.
![]() (I.1)
(I.1)
![]() (I.2)
(I.2)
Уравнение (I.1) является математической записью 1-го начала термодинамики для конечного, уравнение (I.2) – для бесконечно малого изменения состояния системы.
Внутренняя энергия является функцией состояния; это означает, что изменение внутренней энергии ΔU не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутренней энергии U2 и U1 в этих состояниях:
![]() (I.3)
(I.3)
Следует отметить, что определить абсолютное значение внутренней энергии системы невозможно; термодинамику интересует лишь изменение внутренней энергии в ходе какого-либо процесса.
Рассмотрим приложение первого начала термодинамики для определения работы, совершаемой системой при различных термодинамических процессах (мы будем рассматривать простейший случай – работу расширения идеального газа).
Изохорный процесс (V = const; ΔV = 0).
Поскольку работа расширения равна произведению давления и изменения объема, для изохорного процесса получаем:
![]() (I.1)
(I.1)
![]() (I.4)
(I.4)
![]() (I.5)
(I.5)
Изотермический процесс (Т = const).
Из уравнения состояния одного моля идеального газа получаем:
![]() (I.6)
(I.6)
Отсюда:
![]() (I.7)
(I.7)
Проинтегрировав выражение (I.6) от V1 до V2, получим
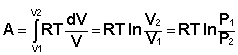 (I.8)
(I.8)
Изобарный процесс (Р = const).
![]() (I.9)
(I.9)
Подставляя полученные выражения для работы различных процессов в уравнение (I.1), для тепловых эффектов этих процессов получим:
![]() (I.10)
(I.10)
![]() (I.11)
(I.11)
![]() (I.12)
(I.12)
В уравнении (I.12) сгруппируем переменные с одинаковыми индексами. Получаем:
![]() (I.13)
(I.13)
Введем новую функцию состояния системы – энтальпию H, тождественно равную сумме внутренней энергии и произведения давления на объем:
![]()
Тогда выражение (I.13) преобразуется к следующему виду:
![]() (I.14)
(I.14)
Т.о., тепловой эффект изобарного процесса равен изменению энтальпии системы.
Адиабатический процесс (Q = 0).
При адиабатическом процессе работа расширения совершается за счёт уменьшения внутренней энергии газа:
![]() (I.15)
(I.15)
В случае если Cv не зависит от температуры (что справедливо для многих реальных газов), работа, произведённая газом при его адиабатическом расширении, прямо пропорциональна разности температур:
![]() (I.16)
(I.16)
1.3 ПРИЛОЖЕНИЯ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
К ХИМИЧЕСКИМ ПРОЦЕССАМ
1.3.1 Закон Гесса
Как известно, большинство химических реакций сопровождаются выделением (экзотермические реакции) либо поглощением (эндотермические реакции) теплоты. Первое начало термодинамики дает возможность рассчитать тепловой эффект химической реакции при различных условиях её проведения.
Тепловой эффект (теплота) химической реакции количество теплоты, выделившейся либо поглотившейся в ходе реакции. Тепловой эффект относят, как правило, к числу молей прореагировавшего исходного вещества, стехиометрический коэффициент перед которым максимален.
Например, реакцию окисления водорода в химической термодинамике записывают в виде:
Н2 + 1/2 О2 ––> Н2О
и тепловой эффект рассчитывают на 1 моль водорода.
Тепловые эффекты, сопровождающие протекание химических реакций, являются предметом одного из разделов химической термодинамики – термохимии. Определим некоторые понятия термохимии.
Теплота образования вещества тепловой эффект реакции образования 1 моля сложного вещества из простых. Теплоты образования простых веществ принимаются равными нулю.
Теплота сгорания вещества тепловой эффект реакции окисления 1 моля вещества в избытке кислорода до высших устойчивых оксидов.
Теплота растворения тепловой эффект процесса растворения 1 моля вещества в бесконечно большом количестве растворителя. Теплота растворения складывается из двух составляющих: теплоты разрушения кристаллической решетки (для твердого вещества) и теплоты сольватации:
![]()
Поскольку ΔНкр.реш всегда положительно (на разрушение кристаллической решетки необходимо затратить энергию), а ΔНсольв всегда отрицательно, знак ΔНраств определяется соотношением абсолютных величин ΔНкр.реш и ΔНсольв:
![]()
Основным законом термохимии является закон Гесса, являющийся частным случаем первого начала термодинамики:
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Выше было показано, что изменение энтальпии ΔН (тепловой эффект изобарного процесса Qp) и изменение внутренней энергии ΔU (тепловой эффект изохорного процесса Qv) не зависят от пути, по которому система переходит из начального состояния в конечное.
Рассмотрим некоторый обобщенный химический процесс превращения исходных веществ А1, А2, А3 в продукты реакции В1, В2, В3, который может быть осуществлен различными путями в одну или несколько стадий:
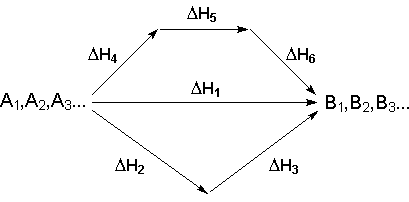
Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:
![]() (I.17)
(I.17)
Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты химических процессов. В термохимических расчетах обычно используют ряд следствий из закона Гесса:
1. Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье – Лапласа).
2. Для двух реакций, имеющих одинаковые исходные, но разные конечные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного конечного состояния в другое.
С + О2 ––> СО + 1/2 О2 ΔН1
С + О2 ––> СО2 ΔН2
СО + 1/2 О2 > СО2 ΔН3
![]() (I.18)
(I.18)
3. Для двух реакций, имеющих одинаковые конечные, но разные исходные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного исходного состояния в другое.
С(алмаз) + О2 > СО2 ΔН1
С(графит) + О2 > СО2 ΔН2
С(алмаз) ––> С(графит) ΔН3
![]() (I.19)
(I.19)
4. Тепловой эффект химической реакции равен разности сумм теплот образования продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты.
![]() (I.20)
(I.20)
5. Тепловой эффект химической реакции равен разности сумм теплот сгорания исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты.
![]() (I.21)
(I.21)
В качестве примера рассмотрим расчет теплового эффекта реакции окисления одного моля глюкозы (теплота образования кислорода по определению равна нулю):
С6Н12О6 + 6 О2 ––> 6 СО2 + 6 Н2О
![]()
Величины тепловых эффектов химических реакций зависят
от условий, в которых проводятся реакции. Поэтому табличные значения теплот
различных процессов принято относить к стандартному состоянию – температуре 298
К и давлению 101325 Па (760 мм. рт. ст.; 1 атм.); величины тепловых эффектов
при данных условиях называют стандартными тепловыми эффектами и обозначают
ΔН°298 и ΔU°298 соответственно.
1.3.2 Зависимость теплового эффекта реакции от температуры. Закон Кирхгофа
В общем случае тепловой эффект химической реакции зависит от температуры и давления, при которых проводится реакция. Влиянием давления на ΔН и ΔU реакции обычно пренебрегают. Влияние температуры на величины тепловых эффектов описывает закон Кирхгофа:
Температурный коэффициент теплового эффекта химической реакции равен изменению теплоемкости системы в ходе реакции.
Продифференцируем ΔН и ΔU по температуре при постоянных давлении и температуре соответственно:
![]() (I.22)
(I.22)
![]() (I.23)
(I.23)
Производные энтальпии и внутренней энергии системы по температуре есть теплоемкости системы в изобарных и изохорных условиях Cp и Cv соответственно:
![]() (I.24)
(I.24)
![]() (I.25)
(I.25)
Подставив выражения (I.24, I.25) в (I.22, I.23), получаем математическую запись закона Кирхгофа:
![]() (I.26)
(I.26)
![]() (I.27)
(I.27)
Для химического процесса изменение теплоемкости задается изменением состава системы и рассчитывается следующим образом:
![]() (I.28)
(I.28)
![]() (I.29)
(I.29)
Если проинтегрировать выражения (I.26, I.27) от Т = Т1 до Т = Т2, считая ΔСp (ΔСv) не зависящим от температуры, получим интегральную форму закона Кирхгофа:
![]() (I.30)
(I.30)
![]() (I.31)
(I.31)
Поскольку обычно известны табличные значения стандартных тепловых эффектов ΔН°298 и ΔU°298, преобразуем выражения (I.30, I.31):
![]() (I.32)
(I.32)
![]() (I.33)
(I.33)
При расчете изменения теплового эффекта реакции в большом интервале температур необходимо учитывать зависимость теплоёмкости от температуры, которая выражается степенным рядом C°p = aT + bT2 + cT3; коэффициенты a, b, c приведены в справочниках.
1.4 ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНТРОПИЯ
Первое начало термодинамики утверждает, что при превращении одной формы энергии в другую полная энергия системы не изменяется, однако не указывает никаких ограничений относительно возможности этого процесса. Поэтому первое начало термодинамики позволяет рассчитать энергетический эффект процесса, однако не дает ответа на вопросы о том, будет ли процесс протекать самопроизвольно, о направлении и глубине протекания процесса.
Самопроизвольный процесс процесс, который может протекать без затраты работы извне, причем в результате может быть получена работа в количестве, пропорциональном произошедшему изменению состояния системы. Самопроизвольный процесс может протекать или обратимо, или необратимо. Хотя определение обратимого процесса уже приводилось, следует подробнее рассмотреть это понятие. Чтобы самопроизвольный процесс протекал обратимо, необходимо приложить извне такое сопротивление, чтобы переход был очень медленным и при бесконечно малом изменении противодействующей силы процесс мог пойти в обратном направлении. В случае обратимо происходящего изменения состояния системы производится максимальное количество работы. Всякий реальный процесс в какой-то степени является необратимым, и получаемая работа меньше максимально возможного теоретического количества.
Вынужденный процесс процесс, для протекания которого требуется затрата работы извне в количестве, пропорциональном производимому изменению состояния системы.
Второе начало термодинамики дает возможность определить, какой из процессов будет протекать самопроизвольно, какое количество работы может быть при этом получено, каков предел самопроизвольного течения процесса. Далее, второе начало термодинамики дает возможность определить, какими должны быть условия, чтобы нужный процесс протекал в необходимом направлении и в требуемой степени, что особенно важно для решения различных задач прикладного характера. Подобно первому, второе начало термодинамики выведено непосредственно из опыта. В то же время второе начало термодинамики имеет ограниченную область применения: оно применимо лишь к макроскопическим системам. Ниже приведены некоторые формулировки второго начала термодинамики:
Теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому (постулат Клаузиуса).
Невозможен процесс, единственным результатом которого является превращение теплоты в работу.
Невозможно построить машину, все действия которой сводились бы к производству работы за счет охлаждения теплового источника (вечный двигатель второго рода).
Рассмотрим работу тепловой машины, т.е. машины, производящей работу за счет теплоты, поглощаемой от какого-либо тела, называемого нагревателем. Нагреватель с температурой Т1 передает теплоту Q1 рабочему телу, например, идеальному газу, совершающему работу расширения А; чтобы вернуться в исходное состояние, рабочее тело должно передать телу, имеющему более низкую температуру Т2 (холодильнику), некоторое количество теплоты Q2, причем
![]() (I.34)
(I.34)
Отношение работы А, совершенной тепловой машиной, к количеству теплоты Q1, полученному от нагревателя, называется термодинамическим коэффициентом полезного действия (КПД) машины η:
![]() (I.35)
(I.35)
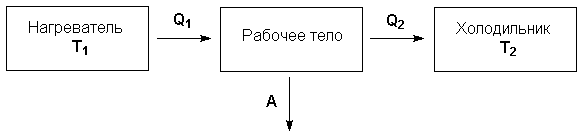
Рисунок 1.1 Схема
тепловой машины
Для получения математического выражения второго начала термодинамики рассмотрим работу идеальной тепловой машины (машины, обратимо работающей без трения и потерь тепла; рабочее тело – идеальный газ). Работа машины основана на принципе обратимого циклического процесса термодинамического цикла Карно (рис. 1.2).
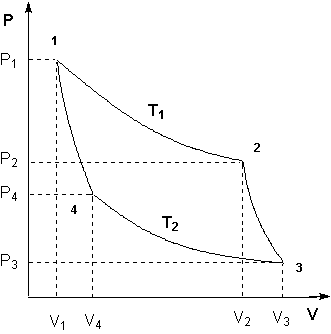
Рисунок 1.2 Цикл Карно
Запишем выражения для работы на всех участках цикла:
Участок 1 – 2: Изотермическое расширение.
![]() (I.36)
(I.36)
Участок 2 – 3: Адиабатическое расширение.
![]() (I.37)
(I.37)
Участок 3 – 4: Изотермическое сжатие.
![]() (I.38)
(I.38)
Участок 4 – 1: Адиабатическое сжатие.
![]() (I.39)
(I.39)
Общая работа в цикле равна сумме работ на всех участках:
![]() (I.40)
(I.40)
Проведя ряд несложных преобразований, получим для КПД идеальной тепловой машины, работающей по циклу Карно:
![]() (I.41)
(I.41)
Т.о., максимальный КПД тепловой машины не зависит от природы рабочего тела, а определяется только разностью температур нагревателя и холодильника. Очевидно, что без перепада температур превращение теплоты в работу невозможно. Полученное выражение справедливо для тепловой машины, обратимо работающей по любому циклу, поскольку любой цикл можно разбить на множество бесконечно малых циклов Карно.
Для необратимо работающей тепловой машины уравнение (I.41) преобразуется в неравенство:
![]() (I.42)
(I.42)
Для общего случая можем записать:
![]() (I.43)
(I.43)
На основе анализа работы идеальной тепловой машины Карно можно сделать следующий вывод, являющийся также одной из формулировок второго начала термодинамики:
Любая форма энергии может полностью перейти в теплоту, но теплота преобразуется в другие формы энергии лишь частично.
Т.о., можно условно принять, что внутренняя энергии системы состоит из двух составляющих: "свободной" X и "связанной" Y энергий, причем "свободная" энергия может быть переведена в работу, а "связанная" энергия может перейти только в теплоту.
![]() (I.44)
(I.44)
Величина связанной энергии тем больше, чем меньше разность температур, и при T = const тепловая машина не может производить работу. Мерой связанной энергии является новая термодинамическая функция состояния, называемая энтропией.
Введем определение энтропии, основываясь на цикле Карно. Преобразуем выражение (I.41) к следующему виду:
![]() (I.45)
(I.45)
Отсюда получаем, что для обратимого цикла Карно отношение количества теплоты к температуре, при которой теплота передана системе (т.н. приведенная теплота) есть величина постоянная:
![]() (I.46)
(I.46) ![]() (I.47)
(I.47)
Это верно для любого обратимого циклического процесса, т.к. его можно представить в виде суммы элементарных циклов Карно, для каждого из которых
![]() (I.48)
(I.48)
Т.о., алгебраическая сумма приведённых теплот для произвольного обратимого цикла равна нулю:
![]() (I.49)
(I.49)
Выражение (I.49) для любого цикла может быть заменено интегралом по замкнутому контуру:
![]() (I.50)
(I.50)
Если интеграл по замкнутому контуру равен нулю, то подынтегральное выражение есть полный дифференциал некоторой функции состояния; эта функция состояния есть энтропия S:
![]() (I.51)
(I.51)
Выражение (I.51) является определением новой функции состояния – энтропии и математической записью второго начала термодинамики для обратимых процессов. Если система обратимо переходит из состояния 1 в состояние 2, изменение энтропии будет равно:
![]() (I.52)
(I.52)
Подставляя (I.51, I.52) в выражения для первого начала термодинамики (I.1, I.2) получим совместное аналитическое выражение двух начал термодинамики для обратимых процессов:
![]() (I.53)
(I.53)
![]() (I.54)
(I.54)
Для необратимых процессов можно записать неравенства:
![]() (I.55)
(I.55)
![]() (I.56)
(I.56)
![]() (I.57)
(I.57)
Т.о., как следует из (I.57), работа обратимого процесса всегда больше, чем того же процесса, проводимого необратимо. Если рассматривать изолированную систему (δQ = 0), то легко показать, что для обратимого процесса dS = 0, а для самопроизвольного необратимого процесса dS > 0.
В изолированных системах самопроизвольно могут протекать только процессы, сопровождающиеся увеличением энтропии.
Энтропия изолированной системы не может самопроизвольно убывать.
Оба этих вывода также являются формулировками второго начала термодинамики.
1.4.1 Статистическая интерпретация энтропии
Классическая термодинамика рассматривает происходящие процессы безотносительно к внутреннему строению системы; поэтому в рамках классической термодинамики показать физический смысл энтропии невозможно. Для решения этой проблемы Больцманом в теорию теплоты были введены статистические представления. Каждому состоянию системы приписывается термодинамическая вероятность (определяемая как число микросостояний, составляющих данное макросостояние системы), тем большая, чем более неупорядоченным или неопределенным является это состояние. Т.о., энтропия есть функция состояния, описывающая степень неупорядоченности системы. Количественная связь между энтропией S и термодинамической вероятностью W выражается формулой Больцмана:
![]() (I.58)
(I.58)
С точки зрения статистической термодинамики второе начало термодинамики можно сформулировать следующим образом:
Система стремится самопроизвольно перейти в состояние с максимальной термодинамической вероятностью.
Статистическое толкование второго начала термодинамики придает энтропии конкретный физический смысл меры термодинамической вероятности состояния системы.
1.5 ТРЕТЬЕ НАЧАЛО ТЕРМОДИНАМИКИ
Ранее мы показали, что внутреннюю энергию системы можно условно представить в виде суммы двух величин "свободной" и "связанной" энергии. Возможность рассчитать величину "свободной" энергии, т.е. той части внутренней энергии системы, которую можно превратить в работу, дает тепловая теорема Нернста, называемая также третьим начало термодинамики.
Основные положения тепловой теоремы заключаются в следующем:
1. При абсолютном нуле температуры свободная энергия X равна теплоте процесса.
![]() (I.59)
(I.59)
2. При температурах, близких к абсолютному нулю, теплоемкость системы равна нулю.
![]() (I.60)
(I.60)
Одной из формулировок третьего начала термодинамики является также постулат Планка:
Энтропия идеального кристалла при абсолютном нуле температуры равна нулю.
Строго говоря, тепловая теорема Нернста и постулат
Планка являются следствиями из второго начала термодинамики; но независимо от
этого они имеют очень большое значение, позволяя рассчитывать абсолютную
энтропию системы и, следовательно, величину свободной энергии системы.
1.5.1 Расчет абсолютной энтропии
Рассчитаем изменение энтропии некоторой системы при нагревании её от абсолютного нуля до температуры T при постоянном давлении. Из первого и второго начал термодинамики имеем:
![]() (I.61)
(I.61)
![]() (I.62)
(I.62)
Отсюда:
![]() (I.63)
(I.63)
Учитывая, что ST=0 = 0, получим:
![]() (I.64)
(I.64)
При T = 0 любое вещество может находиться только в твердом состоянии. При нагревании вещества возможен его переход в жидкое и затем в газообразное состояние; для фазовых переходов, происходящих в изобарно-изотермических условиях, изменение энтропии равно приведенной теплоте фазового перехода:
![]() (I.65)
(I.65)
Таким образом, нагревание вещества без фазовых переходов сопровождается непрерывным ростом энтропии; при фазовом переходе происходит скачкообразное изменение энтропии. Графическая зависимость энтропии вещества от температуры приведена на рисунке 1.3.
Учитывая это, рассчитать абсолютную энтропию любого вещества при любой температуре можно следующим образом:
![]() (I.66)
(I.66)
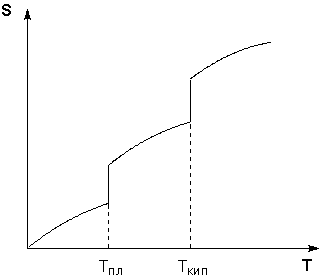
Рис. 1.3 Зависимость энтропии вещества от температуры.
Поскольку энтропия есть функция состояния, изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции; оно может быть рассчитано по уравнению (I.67):
![]() (I.67)
(I.67)
Для многих веществ величины абсолютной энтропии в стандартных условиях приведены в справочной литературе.
1.6 ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ
Изменение энтропии однозначно определяет направление и предел самопроизвольного протекания процесса лишь для наиболее простых систем изолированных. На практике же большей частью приходится иметь дело с системами, взаимодействующими с окружающей средой. Для характеристики процессов, протекающих в закрытых системах, были введены новые термодинамические функции состояния: изобарно-изотермический потенциал (свободная энергия Гиббса) и изохорно-изотермический потенциал (свободная энергия Гельмгольца).
Поведение всякой термодинамической системы в общем случае определяется одновременным действием двух факторов – энтальпийного, отражающего стремление системы к минимуму тепловой энергии, и энтропийного, отражающего противоположную тенденцию – стремление системы к максимальной неупорядоченности. Если для изолированных систем (ΔН = 0) направление и предел самопроизвольного протекания процесса однозначно определяется величиной изменения энтропии системы ΔS, а для систем, находящихся при температурах, близких к абсолютному нулю (S = 0 либо S = const) критерием направленности самопроизвольного процесса является изменение энтальпии ΔН, то для закрытых систем при температурах, не равных нулю, необходимо одновременно учитывать оба фактора. Направлением и предел самопроизвольного протекания процесса в любых системах определяет более общий принцип минимума свободной энергии:
Самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения.
Для закрытых систем, находящихся в изобарно-изотермических либо изохорно-изотермических условиях свободная энергия принимает вид изобарно-изотермического либо изохорно-изотермического потенциалов (т.н. свободная энергия Гиббса и Гельмгольца соответственно). Данные функции называют иногда просто термодинамическими потенциалами, что не вполне строго, поскольку термодинамическими потенциалами являются также внутренняя энергия (изохорно-изэнтропный) и энтальпия (изобарно-изэнтропный потенциал).
Рассмотрим закрытую систему, в которой осуществляется равновесный процесс при постоянных температуре и объеме. Выразим работу данного процесса, которую обозначим Amax (поскольку работа процесса, проводимого равновесно, максимальна), из уравнений (I.53, I.54):
![]() (I.68)
(I.68)
![]() (I.69)
(I.69)
Преобразуем выражение (I.69), сгруппировав члены с одинаковыми индексами:
![]() (I.70)
(I.70)
Введя обозначение:
![]() (I.71)
(I.71)
получаем:
![]() (I.72)
(I.72)
![]() (I.73)
(I.73)
Функция ![]() есть изохорно-изотермический
потенциал (свободная энергия Гельмгольца), определяющий направление и
предел самопроизвольного протекания процесса в закрытой системе, находящейся в
изохорно-изотермических условиях.
есть изохорно-изотермический
потенциал (свободная энергия Гельмгольца), определяющий направление и
предел самопроизвольного протекания процесса в закрытой системе, находящейся в
изохорно-изотермических условиях.
Закрытую систему, находящуюся в изобарно-изотермических условиях, характеризует изобарно-изотермический потенциал G:
![]() (I.74)
(I.74)
![]() (I.75)
(I.75)
Поскольку –ΔF = Amax, можно записать:
![]() (I.76)
(I.76)
Величину А'max называют максимальной
полезной работой (максимальная работа за вычетом работы расширения).
Основываясь на принципе минимума свободной энергии, можно сформулировать
условия самопроизвольного протекания процесса в закрытых системах.
Условия самопроизвольного протекания процессов в закрытых системах:
Изобарно-изотермические (P = const, T = const):
ΔG < 0, dG < 0
ΔF < 0, dF < 0
Процессы, которые сопровождаются увеличением термодинамических потенциалов, протекают лишь при совершении работы извне над системой. В химии наиболее часто используется изобарно-изотермический потенциал, поскольку большинство химических (и биологических) процессов происходят при постоянном давлении. Для химических процессов величину ΔG можно рассчитать, зная ΔH и ΔS процесса, по уравнению (I.75), либо пользуясь таблицами стандартных термодинамических потенциалов образования веществ ΔG°обр; в этом случае ΔG° реакции рассчитывается аналогично ΔН° по уравнению (I.77):
![]() (I.77)
(I.77)
Величина стандартного изменения изобарно-изотермического потенциала в ходе химической любой реакции ΔG°298 есть мера химического сродства исходных веществ. Основываясь на уравнении (I.75), можно оценить вклад энтальпийного и энтропийного факторов в величину ΔG и сделать некоторые обобщающие заключения о возможности самопроизвольного протекания химических процессов, основываясь на знаке величин ΔН и ΔS.
1. Экзотермические реакции; ΔH < 0.
а) Если ΔS > 0, то ΔG всегда отрицательно; экзотермические реакции, сопровождающиеся увеличением энтропии, всегда протекают самопроизвольно.
б) Если ΔS < 0, реакция будет идти самопроизвольно при ΔН > TΔS (низкие температуры).
2. Эндотермические реакции; ΔH > 0.
а) Если ΔS > 0, процесс будет самопроизвольным при ΔН < TΔS (высокие температуры).
б) Если ΔS < 0, то ΔG всегда положительно; самопроизвольное протекание эндотермических реакций, сопровождающихся уменьшением энтропии, невозможно.
1.7 ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Как было показано выше, протекание самопроизвольного процесса в термодинамической системе сопровождается уменьшением свободной энергии системы (dG < 0, dF < 0). Очевидно, что рано или поздно (напомним, что понятие "время" в термодинамике отсутствует) система достигнет минимума свободной энергии. Условием минимума некоторой функции Y = f(x) является равенство нулю первой производной и положительный знак второй производной: dY = 0; d2Y > 0. Таким образом, условием термодинамического равновесия в закрытой системе является минимальное значение соответствующего термодинамического потенциала:
Изобарно-изотермические (P = const, T = const):
ΔG = 0 dG = 0, d2G > 0
Изохорно-изотермические (V = const, T = const):
ΔF = 0 dF = 0, d2F > 0
Состояние системы с минимальной свободной энергией есть состояние термодинамического равновесия:
Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом.
Учение о равновесных состояниях – один из разделов термодинамики. Далее мы будем рассматривать частный случай термодинамического равновесного состояния – химическое равновесие. Как известно, многие химические реакции являются обратимыми, т.е. могут одновременно протекать в обоих направлениях – прямом и обратном. Если проводить обратимую реакцию в закрытой системе, то через некоторое время система придет в состояние химического равновесия – концентрации всех реагирующих веществ перестанут изменяться во времени. Необходимо отметить, что достижение системой состояния равновесия не означает прекращения процесса; химическое равновесие является динамическим, т.е. соответствует одновременному протеканию процесса в противоположных направлениях с одинаковой скоростью. Химическое равновесие является подвижным всякое бесконечно малое внешнее воздействие на равновесную систему вызывает бесконечно малое изменение состояния системы; по прекращении внешнего воздействия система возвращается в исходное состояние. Ещё одним важным свойством химического равновесия является то, что система может самопроизвольно прийти в состояние равновесия с двух противоположных сторон. Иначе говоря, любое состояние, смежное с равновесным, является менее устойчивым, и переход в него из состояния равновесия всегда связан с необходимостью затраты работы извне.
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции
![]()
соответствующие константы равновесия выражаются следующим образом:
![]() (I.78)
(I.78) ![]() (I.79)
(I.79)
![]() (I.80)
(I.80)
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений.
Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
![]() (I.81)
(I.81)
![]() (I.82)
(I.82)
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие.
Приняв, что V1 = V2, можно записать:
![]() (I.83)
(I.83)
![]() (I.84)
(I.84)
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л.
Теперь рассмотрим (с некоторыми упрощениями) более строгий термодинамический вывод выражения для константы равновесия. Для этого необходимо ввести понятие химический потенциал. Очевидно, что величина свободной энергии системы будет зависеть как от внешних условий (T, P или V), так и от природы и количества веществ, составляющих систему. В случае, если состав системы изменяется во времени (т.е. в системе протекает химическая реакция), необходимо учесть влияние изменения состава на величину свободной энергии системы. Введем в некоторую систему бесконечно малое количество dni молей i-го компонента; это вызовет бесконечно малое изменение термодинамического потенциала системы. Отношение бесконечно малого изменения величины свободной энергии системы к бесконечно малому количеству компонента, внесенному в систему, есть химический потенциал μi данного компонента в системе:
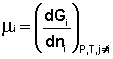 (I.85)
(I.85)
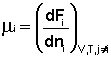 (I.86)
(I.86)
Химический потенциал компонента связан с его парциальным давлением или концентрацией следующими соотношениями:
![]() (I.87)
(I.87)
![]() (I.88)
(I.88)
Здесь μ°i – стандартный химический потенциал компонента (Pi = 1 атм., Сi = 1 моль/л.). Очевидно, что изменение свободной энергии системы можно связать с изменением состава системы следующим образом:
![]() (I.89)
(I.89)
![]() (I.90)
(I.90)
Поскольку условием равновесия является минимум свободной энергии системы (dG = 0, dF = 0), можно записать:
![]() (I.91)
(I.91)
В закрытой системе изменение числа молей одного компонента сопровождается эквивалентным изменением числа молей остальных компонентов; т.е., для приведенной выше химической реакции имеет место соотношение:
![]() (I.92)
(I.92)
Отсюда можно получить следующее условие химического равновесия в закрытой системе:
![]() (I.93)
(I.93)
В общем виде условие химического равновесия можно записать следующим образом:
![]() (I.94)
(I.94)
Выражение (I.94) носит название уравнения Гиббса – Дюгема. Подставив в него зависимость химического потенциала от концентрации, получаем:
![]() (I.95)
(I.95)
Поскольку Σniμi = ΔF, а Σniμ°i = ΔF°, получаем:
![]() (I.96)
(I.96)
Для изобарно-изотермического процесса аналогичным образом можно получить:
![]() (I.97)
(I.97)
Полученные нами выражения I.96 – I.97 есть изотерма химической реакции. Если система находится в состоянии химического равновесия, то изменение термодинамического потенциала равно нулю; получаем:
![]() (I.98)
(I.98)
![]() (I.99)
(I.99)
Здесь сi и рi – равновесные концентрации и парциальные давления исходных веществ и продуктов реакции (в отличие от неравновесных Сi и Рi в уравнениях I.96 I.97).
Поскольку для каждой химической реакции стандартное изменение термодинамического потенциала ΔF° и ΔG° есть строго определенная величина, то произведение равновесных парциальных давлений (концентраций), возведенных в степень, равную стехиометрическому коэффициенту при данном веществе в уравнении химической реакции (стехиометрические коэффициенты при исходных веществах принято считать отрицательными) есть некоторая константа, называемая константой равновесия. Уравнения (I.98, I.99) показывают связь константы равновесия со стандартным изменением свободной энергии в ходе реакции. Уравнение изотермы химической реакции связывает величины реальных концентраций (давлений) реагентов в системе, стандартного изменения термодинамического потенциала в ходе реакции и изменения термодинамического потенциала при переходе из данного состояния системы в равновесное. Знак ΔG (ΔF) определяет возможность самопроизвольного протекания процесса в системе. При этом ΔG° (ΔF°) равно изменению свободной энергии системы при переходе из стандартного состояния (Pi = 1 атм., Сi = 1 моль/л) в равновесное. Уравнение изотермы химической реакции позволяет рассчитать величину ΔG (ΔF) при переходе из любого состояния системы в равновесное, т.е. ответить на вопрос, будет ли химическая реакция протекать самопроизвольно при данных концентрациях Сi (давлениях Рi) реагентов:
![]() (I.100)
(I.100)
![]() (I.101)
(I.101)
Если изменение термодинамического потенциала меньше
нуля, процесс в данных условиях будет протекать самопроизвольно.
1.7.1 Влияние внешних условий на химическое равновесие
При постоянстве внешних условий система может находиться в состоянии равновесия сколь угодно долго. Если изменить эти условия (т.е. оказать на систему какое-либо внешнее воздействие), равновесие нарушается; в системе возникает самопроизвольный процесс, который продолжается до тех пор, пока система опять не достигнет состояния равновесия (уже при новых условиях). Рассмотрим, как влияют на положение равновесия некоторые факторы.
1.7.2 Влияние давления и концентрации
Рассмотрим несколько возможных случаев смещения равновесия.
1. В систему добавлено исходное вещество. В этом случае
![]() ;
; ![]() ;
;
По уравнению изотермы химической реакции (I.100 I.101) получаем: ΔF < 0; ΔG < 0. В системе возникнет самопроизвольный химический процесс, направленный в сторону расходования исходных веществ и образования продуктов реакции (химическое равновесие смещается вправо).
2. В систему добавлен продукт реакции. В этом случае
![]() ;
; ![]() ;
;
Согласно уравнению изотермы химической реакции, ΔF > 0; ΔG > 0. Химическое равновесие будет смещено влево (в сторону расходования продуктов реакции и образования исходных веществ).
3. Изменено общее давление (для реакций в газовой фазе).
Парциальные давления всех компонентов Рi в этом случае изменяются в одинаковой степени; направление смещения равновесия будет определяться суммой стехиометрических коэффициентов Δn.
Учитывая, что парциальное давление газа в смеси равно общему давлению, умноженному на мольную долю компонента в смеси (Рi = РХi), изотерму реакции можно переписать в следующем виде (здесь Δn = Σ(ni)прод – Σ(ni)исх):
![]() (I.102)
(I.102)
![]() (I.103)
(I.103)
Примем, что Р2 > Р1. В этом случае, если Δn > 0 (реакция идет с увеличением числа молей газообразных веществ), то ΔG > 0; равновесие смещается влево. Если реакция идет с уменьшением числа молей газообразных веществ (Δn < 0), то ΔG < 0; равновесие смещается вправо. Иначе говоря, увеличение общего давления смещает равновесие в сторону процесса, идущего с уменьшением числа молей газообразных веществ. Уменьшение общего давления газов в смеси (Р2 < Р1) будет смещать равновесие в сторону реакции, идущей с увеличением числа молей газообразных веществ.
Необходимо отметить, что изменение концентрации или
давления, смещая равновесие, не изменяет величину константы равновесия, которая
зависит только от природы реагирующих веществ и температуры.
1.7.3 Влияние температуры на положение равновесия
Повышение либо понижение температуры означает приобретение либо потерю системой энергии и, следовательно, должно изменять величину константы равновесия.
Запишем уравнение (I.99) в следующем виде:
![]() (I.104)
(I.104)
![]() (I.105)
(I.105)
Продифференцировав выражение (I.105) по температуре, получаем для зависимости константы равновесия от температуры уравнение (I.106) изобару Вант-Гоффа:
![]() (I.06)
(I.06)
Рассуждая аналогичным образом, для процесса, проходящего в изохорных условиях, можно получить изохору Вант-Гоффа:
![]() (I.107)
(I.107)
Изобара и изохора Вант-Гоффа связывают изменение константы химического равновесия с тепловым эффектом реакции в изобарных и изохорных условиях соответственно. Очевидно, что чем больше по абсолютной величине тепловой эффект химической реакции, тем сильнее влияет температура на величину константы равновесия. Если реакция не сопровождается тепловым эффектом, то константа равновесия не зависит от температуры.
Экзотермические реакции: ΔH° < 0 (ΔU° < 0). В этом случае, согласно (I.106, I.107), температурный коэффициент логарифма константы равновесия отрицателен. Повышение температуры уменьшает величину константы равновесия, т.е. смещает равновесие влево.
Эндотермические реакции: ΔH° > 0 (ΔU° > 0). В этом случае температурный коэффициент логарифма константы равновесия положителен; повышение температуры увеличивает величину константы равновесия (смещает равновесие вправо).
Графики зависимостей константы равновесия от
температуры для экзотермических и эндотермических реакций приведены на рис.
I.4.
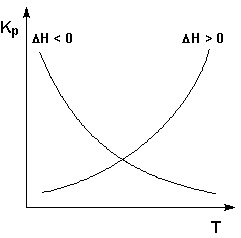
Рис. 1.4 Зависимость
константы равновесия от температуры.
Действие рассмотренных нами факторов (давления, концентрации и температуры), равно как и любых других, на систему, находящуюся в состоянии равновесия, обобщает принцип смещения равновесия, называемый также принципом Ле Шателье – Брауна:
Если на систему, находящуюся в состоянии истинного равновесия, оказывается внешнее воздействие, то в системе возникает самопроизвольный процесс, компенсирующий данное воздействие.
Принцип Ле Шателье – Брауна является одним из следствий второго начала термодинамики и применим к любым макроскопическим системам, находящимся в состоянии истинного равновесия.
1.8 ФАЗОВЫЕ РАВНОВЕСИЯ
Вещество при изменении давления и температуры может переходить из одного агрегатного состояния в другое. Эти переходы, совершающиеся при постоянной температуре, называют фазовыми переходами первого рода. Количество теплоты, которое вещество получает из окружающей среды либо отдает окружающей среде при фазовом переходе, есть скрытая теплота фазового перехода λфп. Если рассматривается гетерогенная система, в которой нет химических взаимодействий, а возможны лишь фазовые переходы, то при постоянстве температуры и давления в системе существует т.н. фазовое равновесие. Фазовое равновесие характеризуется некоторым числом фаз, компонентов и числом степеней термодинамической свободы системы.
Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне её. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними. Число степеней свободы число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе.
Число степеней свободы гетерогенной термодинамической системы, находящейся в состоянии фазового равновесия, определяется правилом фаз, сформулированным Дж. Гиббсом:
Число степеней свободы равновесной термодинамической системы С равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие.
Для системы, на которую из внешних факторов влияют только температура и давление, можно записать:
С = К – Ф + 2 (I.108)
Системы принято классифицировать по числу компонентов (одно-, двухкомпонентные и т.д.), по числу фаз (одно-, двухфазные и т.д.) и числу степеней свободы (инвариантные, моно-, дивариантные и т.д.). Для систем с фазовыми переходами обычно рассматривают графическую зависимость состояния системы от внешних условий – т.н. диаграммы состояния.
Анализ диаграмм состояния позволяет определить число фаз в системе, границы их существования, характер взаимодействия компонентов. В основе анализа диаграмм состояния лежат два принципа: принцип непрерывности и принцип соответствия. Согласно принципу непрерывности, при непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в системе, что приводит к скачкообразному изменению свойств системы. Согласно принципу соответствия, на диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т. н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния.
Рассмотрим и проанализируем диаграмму состояния воды (рис.1.4). Поскольку вода – единственное присутствующее в системе вещество, число независимых компонентов К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА – зависимость давления насыщенного пара воды от температуры), твердым телом и газом (линия ОВ – зависимость давления насыщенного пара надо льдом от температуры), твердым телом и жидкостью (линия ОС – зависимость температуры плавления льда от давления). Три кривые имеют точку пересечения О, называемую тройной точкой воды; тройная точка отвечает равновесию между тремя фазами.
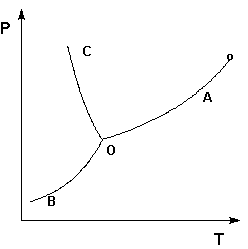
Рис. 1.4. Диаграмма состояния воды
В тройной точке система трехфазна и число степеней свободы равно нулю; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды тройная точка отвечает состоянию с Р = 6.1 кПа и Т = 273.16 К).
Кривая ОВ теоретически продолжается до абсолютного нуля, а кривая давления насыщенного пара над жидкостью ОА заканчивается в критической точке воды (Tкр = 607.46 К, Ркр = 19.5 МПа); выше критической температуры газ и жидкость не могут существовать как отдельные фазы. Кривая ОС в верхней части (при высоких давлениях) изменяет свой наклон (появляются новые кристаллические фазы, плотность которых, в отличие от обычного льда, выше, чем у воды).
Внутри каждой из областей диаграммы (АОВ, ВОС, АОС) система однофазна; число степеней свободы системы равно двум (система дивариантна), т.е. можно одновременно изменять и температуру, и давление, не вызывая изменения числа фаз в системе:
С = 1 – 1 + 2 = 2
На каждой из линий число фаз в системе равно двум и, согласно правилу фаз, система моновариантна, т.е. для каждого значения температуры имеется только одно значение давления, при котором система двухфазна:
С = 1 – 2 + 2 = 1
Влияние давления на температуру фазового перехода описывает уравнение Клаузиуса – Клапейрона:
![]() (I.109)
(I.109)
Здесь ΔVфп = V2 – V1 есть изменение молярного объема вещества при фазовом переходе (причем V2 относится к состоянию, переход в которое сопровождается поглощением теплоты). Уравнение Клаузиуса – Клапейрона позволяет объяснить наклон кривых равновесия на диаграмме состояния однокомпонентной системы. Для переходов "жидкость пар" и "твердое вещество – пар" ΔV всегда больше нуля; поэтому кривые на диаграмме состояния, отвечающие этим равновесиям, всегда наклонены вправо (повышение температуры всегда увеличивает давление насыщенного пара). Поскольку молярный объем газа много больше молярного объема того же вещества в жидком или твердом агрегатном состояниях (Vг >> Vж, Vг >> Vт), уравнение (I.109) для частных случаев испарения и возгонки примет следующий вид:
![]() (I.110)
(I.110)
Для многих веществ скрытая теплота парообразования или возгонки постоянна в большом интервале температур; в этом случае уравнение (I.110) можно проинтегрировать:
![]() (I.111)
(I.111)
Кривая равновесия "твердое вещество жидкость" на диаграммах состояния воды и висмута наклонена влево, а на диаграммах состояния остальных веществ – вправо. Это связано с тем, что плотность воды больше, чем плотность льда (и плотность жидкого висмута больше его плотности в твердом состоянии), т.е. плавление сопровождается уменьшением объема (ΔV < 0). Как следует из выражения (I.111), в этом случае увеличение давления будет понижать температуру фазового перехода "твердое тело – жидкость" (воду и висмут относят поэтому к т.н. аномальным веществам). Для всех остальных веществ (т.н. нормальные вещества) ΔVпл > 0 и, согласно уравнению Клаузиуса Клапейрона, увеличение давления приводит к повышению температуры плавления.
2 ХИМИЧЕСКАЯ КИНЕТИКА
Законы химической термодинамики позволяют определить направление и предел протекания возможного при данных условиях химического процесса, а также его энергетический эффект. Однако термодинамика не может ответить на вопросы о том, как осуществляется данный процесс и с какой скоростью. Эти вопросы – механизм и скорость химической реакции – и являются предметом химической кинетики.
2.1 Скорость химической реакции
Дадим определение основному понятию химической кинетики – скорости химической реакции:
Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).
Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени.
Первое определение является наиболее строгим; из него следует, что скорость химической реакции можно также выражать как изменение во времени любого параметра состояния системы, зависящего от числа частиц какого-либо реагирующего вещества, отнесенное к единице объема или поверхности – электропроводности, оптической плотности, диэлектрической проницаемости и т.д. и т.п. Однако наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. В случае односторонних (необратимых) химических реакций (здесь и далее рассматриваются только односторонние реакции) очевидно, что концентрации исходных веществ во времени постоянно уменьшаются (ΔСисх < 0), а концентрации продуктов реакции увеличиваются (ΔСпрод > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δt записывается следующим образом:
![]() (II.1)
(II.1)
В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
![]() (II.2)
(II.2)
Графическое изображение зависимости концентрации
реагентов от времени есть кинетическая кривая (рисунок 2.1).
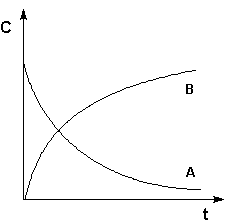
Рис. 2.1 Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой (рис. 2.2); истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:
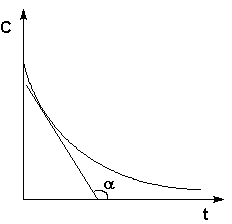
Рис. 2.2 Графическое определение Vист.
![]() (II.3)
(II.3)
Необходимо отметить, что в том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось. Очевидно, что в реакции
2Н2 + О2 ––> 2Н2О
концентрации водорода, кислорода и воды изменяются в
различной степени:
ΔС(Н2) = ΔС(Н2О) = 2 ΔС(О2).
Скорость химической реакции зависит от множества факторов: природы реагирующих веществ, их концентрации, температуры, природы растворителя и т.д.
2.1.1 Кинетическое уравнение химической реакции. Порядок реакции.
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях.
Т. е. для реакции
аА + bВ + dD + ... ––> еЕ + ...
можно записать:
![]() (II.4)
(II.4)
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (II.4) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени.
В химической кинетике принято классифицировать реакции по величине общего порядка реакции. Рассмотрим зависимость концентрации реагирующих веществ от времени для необратимых (односторонних) реакций нулевого, первого и второго порядков.
2.1.2 Реакции нулевого порядка
Для реакций нулевого порядка кинетическое уравнение имеет следующий вид:
![]() (II.5)
(II.5)
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения.
2.1.3 Реакции первого порядка
Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А ––> В. Реакции первого порядка характеризуются кинетическим уравнением вида (II.6). Подставим в него выражение (II.2):
![]() (II.6)
(II.6)
![]() (II.7)
(II.7)
После интегрирования выражения (II.7) получаем:
![]() (II.8)
(II.8)
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации Со. Отсюда следует, что g = ln Со. Получаем:
![]() (II.9)
(II.9)
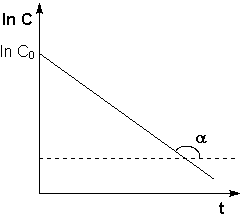
Рис. 2.3 Зависимость логарифма концентрации от времени для реакций первого порядка
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени (рис. 2.3) и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
![]() (II.10)
(II.10)
Из уравнения (II.9) легко получить выражение для константы скорости односторонней реакции первого порядка:
![]() (II.11)
(II.11)
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½Со:
![]() (II.12)
(II.12)
Отсюда
![]() (II.13)
(II.13)
Как видно из полученного выражения, период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества.
2.1.4 Реакции второго порядка
Для реакций второго порядка кинетическое уравнение имеет следующий вид:
![]() (II.14)
(II.14)
либо
![]() (II.15)
(II.15)
Рассмотрим простейший случай, когда кинетическое уравнение имеет вид (II.14) или, что то же самое, в уравнении вида (II.15) концентрации исходных веществ одинаковы; уравнение (II.14) в этом случае можно переписать следующим образом:
![]() (II.16)
(II.16)
После разделения переменных и интегрирования получаем:
![]() (II.17)
(II.17)
Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим:
![]() (II.18)
(II.18)
Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (II.14), характерна линейная зависимость обратной концентрации от времени (рис. 2.4) и константа скорости равна тангенсу угла наклона прямой к оси времени:
![]() (II.19)
(II.19)
![]() (II.20)
(II.20)
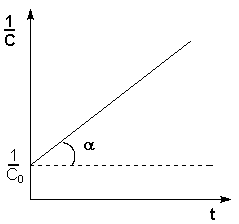
Рис. 2.4 Зависимость обратной концентрации от времени для реакций второго порядка
Если начальные концентрации реагирующих веществ Cо,А и Cо,В различны, то константу скорости реакции находят интегрированием уравнения (II.21), в котором CА и CВ концентрации реагирующих веществ в момент времени t от начала реакции:
![]() (II.21)
(II.21)
В этом случае для константы скорости получаем выражение
![]() (II.22)
(II.22)
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции. Рассмотрим в качестве примера элементарную реакцию гидролиза этилового эфира уксусной кислоты (этилацетата), кинетика которой изучается в лабораторном практикуме по физической химии:
СН3СООС2Н5 + Н2О ––> СН3СООН + С2Н5ОН
Если проводить эту реакцию при близких концентрациях этилацетата и воды, то общий порядок реакции равен двум и кинетическое уравнение имеет следующий вид:
![]() (II.23)
(II.23)
При проведении этой же реакции в условиях большого избытка одного из реагентов (воды или этилацетата) концентрация вещества, находящегося в избытке, практически не изменяется и может быть включена в константу скорости; кинетическое уравнение для двух возможных случаев принимает следующий вид:
1) Избыток воды:
![]() (II.24)
(II.24)
![]() (II.25)
(II.25)
2) Избыток этилацетата:
![]() (II.26)
(II.26)
![]() (II.27)
(II.27)
В этих случаях мы имеем дело с т.н. реакцией псевдопервого порядка. Проведение реакции при большом избытке одного из исходных веществ используется для определения частных порядков реакции.
2.1.5 Методы определения порядка реакции
Проведение реакции в условиях, когда концентрация одного из реагентов много меньше концентрации другого (других) и скорость реакции зависит от концентрации только этого реагента, используется для определения частных порядков реакции – это т.н. метод избыточных концентраций или метод изолирования Оствальда. Порядок реакции по данному веществу определяется одним из перечисленных ниже методов.
Графический метод заключается в
построении графика зависимости концентрации реагента от времени в различных
координатах. Для различных частных порядков эти зависимости имеют следующий
вид:
| Порядок реакции | Зависимость концентрации от времени |
| 1 |
|
| 2 |
|
| 3 |
|
Если построить графики этих зависимостей на основании опытных данных, то лишь одна из них будет являться прямой линией. Если, например, график, построенный по опытным данным, оказался прямолинейным к координатах lnC = f(t), то частный порядок реакции по данному веществу равен единице.
Метод подбора кинетического уравнения заключается в подстановке экспериментальных данных изучения зависимости концентрации вещества от времени в кинетические уравнения различных порядков. Подставляя в приведённые в таблице уравнения значения концентрации реагента в разные моменты времени, вычисляют значения константы скорости. Частный порядок реакции по данному веществу равен порядку того кинетического уравнения, для которого величина константы скорости остаётся постоянной во времени.
| Порядок реакции | Выражение для константы скорости |
| 1 |
|
| 2 |
|
| 3 |
|
Метод определения времени полупревращения
заключается в определении t1/2 для нескольких начальных
концентраций. Как видно из приведённых в таблице уравнений, для реакции первого
порядка время полупревращения не зависит от Co, для реакции второго
порядка – обратно пропорционально Co, и для реакции третьего порядка
обратно пропорционально квадрату начальной концентрации.
| Порядок реакции | Выражение для периода полупревращения |
| 1 |
|
| 2 |
|
| 3 |
|
По характеру зависимости t1/2 от Co нетрудно сделать вывод о порядке реакции по данному веществу. Данный метод, в отличие от описанных выше, применим и для определения дробных порядков.
2.1.6 Молекулярность элементарных реакций
Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности:
Молекулярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия.
Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.):
I2 ––> I + I•
Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных):
СН3Вr + КОН ––> СН3ОН + КВr
Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц:
О2 + NО + NО > 2NО2
Реакции с молекулярностью более трёх неизвестны.
Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Тем не менее, никакой чётко определенной взаимосвязи между понятиями молекулярности и порядка реакции не существует, поскольку порядок реакции характеризует кинетическое уравнение реакции, а молекулярность – механизм реакции.
2.1.7 Сложные реакции
Сложными называют химические реакции, протекающие более чем в одну стадию. Рассмотрим в качестве примера одну из сложных реакций, кинетика и механизм которой хорошо изучены:
2НI + Н2О2 > I2 + 2Н2О
Данная реакция является реакцией второго порядка; е кинетическое уравнение имеет следующий вид:
![]() (II.28)
(II.28)
Изучение механизма реакции показало, что она является двухстадийной (протекает в две стадии):
1) НI + Н2О2 > НIО + Н2О
2) НIО + НI ––> I2 + Н2О
Скорость первой стадии V1 много больше скорости второй стадии V2 и общая скорость реакции определяется скоростью более медленной стадии, называемой поэтому скоростьопределяющей или лимитирующей.
Сделать вывод о том, является реакция элементарной или сложной, можно на основании результатов изучения её кинетики. Реакция является сложной, если экспериментально определенные частные порядки реакции не совпадают с коэффициентами при исходных веществах в стехиометрическом уравнении реакции; частные порядки сложной реакции могут быть дробными либо отрицательными, в кинетическое уравнение сложной реакции могут входить концентрации не только исходных веществ, но и продуктов реакции.
2.1.8 Классификация сложных реакций
Последовательные реакции.
Последовательными называются сложные реакции, протекающие таким образом, что вещества, образующиеся в результате одной стадии (т.е. продукты этой стадии), являются исходными веществами для другой стадии. Схематически последовательную реакцию можно изобразить следующим образом:
А ––> В ––> С ––> ...
Число стадий и веществ, принимающих участие в каждой из стадий, может быть различным.
Параллельные реакции.
Параллельными называют химические реакции, в которых одни и те же исходные вещества одновременно могут образовывать различные продукты реакции, например, два или более изомера:
Сопряжённые реакции.
Сопряжёнными принято называть сложные реакции, протекающие следующим образом:
1) А + В ––> С
2) А + D ––> Е,
причём одна из реакций может протекать самостоятельно, а вторая возможна только при наличии первой. Вещество А, общее для обеих реакций, носит название актор, вещество В – индуктор, вещество D, взаимодействующее с А только при наличии первой реакции – акцептор. Например, бензол в водном растворе не окисляется пероксидом водорода, но при добавлении солей Fe(II) происходит превращение его в фенол и дифенил. Механизм реакции следующий. На первой стадии образуются свободные радикалы:
Fe2+ + H2O2 > Fe3+ + OH– + OH•
которые реагируют с ионами Fe2+ и бензолом:
Fe2+ + OH• ––> Fe3+ + OH–
C6H6 + OH• ––> C6H5• + H2O
Происходит также рекомбинация радикалов:
C6H5 + OH• ––> C6H5ОН
C6H5 + C6H5• ––> C6H5–C6H5
Т.о., обе реакции протекают с участием общего промежуточного свободного радикала OH•.
Цепные реакции.
Цепными называют реакции, состоящие из ряда взаимосвязанных стадий, когда частицы, образующиеся в результате каждой стадии, генерируют последующие стадии. Как правило, цепные реакции протекают с участием свободных радикалов. Для всех цепных реакций характерны три типичные стадии, которые мы рассмотрим на примере фотохимической реакции образования хлороводорода.
1. Зарождение цепи (инициация):
Сl2 + hν ––> 2 Сl•
2. Развитие цепи:
Н2 + Сl• ––> НСl + Н•
Н• + Сl2 ––> НСl + Сl•
Стадия развития цепи характеризуется числом молекул продукта реакции, приходящихся на одну активную частицу – длиной цепи.
3. Обрыв цепи (рекомбинация):
Н• + Н• ––> Н2
Сl• + Сl• ––> Сl2
Н• + Сl• ––> НСl
Обрыв цепи возможен также при взаимодействии активных частиц с материалом стенки сосуда, в котором проводится реакция, поэтому скорость цепных реакций может зависеть от материала и даже от формы реакционного сосуда.
Реакция образования хлороводорода является примером неразветвленной цепной реакции – реакции, в которой на одну прореагировавшую активную частицу приходится не более одной вновь возникающей. Разветвленными называют цепные реакции, в которых на каждую прореагировавшую активную частицу приходится более одной вновь возникающей, т.е. число активных частиц в ходе реакции постоянно возрастает. Примером разветвленной цепной реакции является реакция взаимодействия водорода с кислородом:
1. Инициация:
Н2 + О2 > Н2О + О•
2. Развитие цепи:
О• + Н2 ––> Н• + ОН•
Н• + О2 ––> О• + ОН•
ОН• + Н2 ––> Н2О + Н•
2.1.9 Влияние температуры на константу скорости реакции
Константа скорости реакции есть функция от температуры; повышение температуры, как правило, увеличивает константу скорости. Первая попытка учесть влияние температуры была сделана Вант-Гоффом, который сформулировал следующее эмпирическое правило:
При повышении температуры на каждые 10 градусов константа скорости элементарной химической реакции увеличивается в 2 – 4 раза.
Величина, показывающая, во сколько раз увеличивается константа скорости при повышении температуры на 10 градусов, есть температурный коэффициент константы скорости реакции γ. Математически правило Вант-Гоффа можно записать следующим образом:
![]() (II.29)
(II.29)
![]() (II.30)
(II.30)
Однако правило Вант-Гоффа применимо лишь в узком температурном интервале, поскольку температурный коэффициент скорости реакции γ сам является функцией от температуры; при очень высоких и очень низких температурах γ становится равным единице (т.е. скорость химической реакции перестает зависеть от температуры).
2.1.10 Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Рассмотрим путь некоторой элементарной реакции
А + В ––> С
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом:
А ––> K# ––> B
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'А выше, нежели энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 2.5).
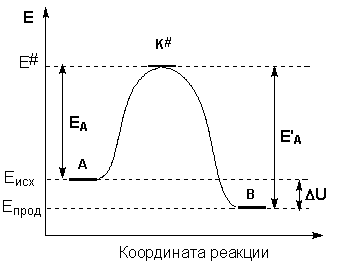
Рис. 2.5 Энергетическая диаграмма химической реакции.
Eисх – средняя энергия частиц исходных веществ,
Eпрод – средняя энергия частиц продуктов реакции
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.6):
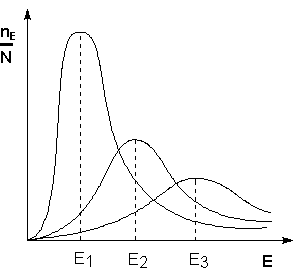
Рис. 2.6 Распределение частиц
по энергии Здесь nЕ/N – доля частиц, обладающих энергией E; Ei
- средняя энергия частиц при температуре Ti (T1 < T2 < T3)
Рассмотрим термодинамический вывод выражения, описывающего зависимость константы скорости реакции от температуры и величины энергии активации – уравнения Аррениуса. Согласно уравнению изобары Вант-Гоффа,
![]() (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
![]() (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
![]() (II.33)
(II.33)
![]() (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации:
![]() (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
![]() (II.36)
(II.36)
![]() (II.37)
(II.37)
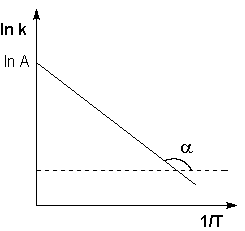
Рис. 2.7 Зависимость логарифма константы скорости химической реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
![]() (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
![]() (II.39)
(II.39)
2.1.11 Кинетика двусторонних (обратимых) реакций
Химические реакции часто являются двусторонними (обратимыми), т.е. могут протекать при данных условиях в двух противоположных направлениях (понятие обратимая реакция следует отличать от термодинамического понятия обратимый процесс; двусторонняя реакция обратима в термодинамическом смысле лишь в состоянии химического равновесия). Рассмотрим элементарную двустороннюю реакцию
А + В <––> D + E
Скорость уменьшения концентрации вещества А при протекании прямой реакции определяется уравнением (II.40):
![]() , (II.40)
, (II.40)
а скорость возрастания концентрации вещества А в результате протекания обратной реакции – уравнением (II.41):
![]() (II.41)
(II.41)
Общая скорость двусторонней реакции в любой момент времени равна разности скоростей прямой и обратной реакции:
![]() (II.42)
(II.42)
По мере протекания двусторонней реакции скорость прямой реакции уменьшается, скорость обратной реакции – увеличивается; в некоторый момент времени скорости прямой и обратной реакции становятся равными и концентрации реагентов перестают изменяться. Таким образом, в результате протекания в закрытой системе двусторонней реакции система достигает состояния химического равновесия; при этом константа равновесия будет равна отношению констант скоростей прямой и обратной реакции:
![]() (II.43)
(II.43)
2.1.12 Кинетика гетерогенных химических реакций
Когда реакция совершается между веществами, находящимися в разных фазах гетерогенной системы, основной постулат химической кинетики становится неприменимым. В гетерогенных реакциях роль промежуточных продуктов обычно играют молекулы, связанные химическими силами с поверхностью раздела фаз (химически адсорбированные на поверхности). Во всяком гетерогенном химическом процессе можно выделить следующие стадии:
1. Диффузия реагентов к реакционной зоне, находящейся на поверхности раздела фаз.
2. Активированная адсорбция частиц реагентов на поверхности.
3. Химическое превращение адсорбированных частиц.
4. Десорбция образовавшихся продуктов реакции.
5. Диффузия продуктов реакции из реакционной зоны.
Стадии 1 и 5 называются диффузионными, стадии 2, 3 и 4 – кинетическими. Универсального выражения для скорости гетерогенных химических реакций не существует, поскольку каждая из выделенных стадий может являться лимитирующей. Как правило, при низких температурах скорость гетерогенной реакции определяют кинетические стадии (т.н. кинетическая область гетерогенного процесса; скорость реакции в этом случае сильно зависит от температуры и величины площади поверхности раздела фаз; порядок реакции при этом может быть любым). При высоких температурах скорость процесса будет определяться скоростью диффузии (диффузионная область гетерогенной реакции, характеризующаяся, как правило, первым порядком реакции и слабой зависимостью скорости процесса от температуры и площади поверхности раздела фаз).
2.2 ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Передача энергии для активации вступающих во взаимодействие молекул может осуществляться либо в форме теплоты (т. н. темновые реакции), либо в виде квантов электромагнитного излучения. Реакции, в которых активация частиц является результатом их взаимодействия с квантами электромагнитного излучения видимой области спектра, называют фотохимическими реакциями. При всех фотохимических процессах выполняется закон Гротгуса:
Химическое превращение вещества может вызвать только то излучение, которое поглощается этим веществом.
Излучение, отражённое веществом, а также прошедшее сквозь него, не вызывают никаких химических превращений. Иногда фотохимические процессы происходят под действием излучения, которое не поглощается реагирующими веществами; однако в таких случаях реакционная смесь должна содержать т.н. сенсибилизаторы. Механизм действия сенсибилизаторов заключается в том, что они поглощают свет, переходя в возбуждённое состояние, а затем при столкновении с молекулами реагентов передают им избыток своей энергии. Сенсибилизатором фотохимических реакций является, например, хлорофилл (см. ниже).
Взаимодействие света с веществом может идти по трём возможным направлениям:
1. Возбуждение частиц (переход электронов на вышележащие орбитали):
A + hν ––> A*
2. Ионизация частиц за счет отрыва электронов:
A + hν ––> A+ + e–
3. Диссоциация молекул с образованием свободных радикалов (гомолитическая) либо ионов (гетеролитическая):
AB + hν > A• + B•
AB + hν > A+ + B–
Между количеством лучистой энергии, поглощенной молекулами вещества, и количеством фотохимически прореагировавших молекул существует соотношение, выражаемое законом фотохимической эквивалентности Штарка Эйнштейна:
Число молекул, подвергшихся первичному фотохимическому превращению, равно числу поглощенных веществом квантов электромагнитного излучения.
Поскольку фотохимическая реакция, как правило, включает в себя и т.н. вторичные процессы (например, в случае цепного механизма), для описания реакции вводится понятие квантовый выход фотохимической реакции:
Квантовый выход фотохимической реакции γ есть отношение числа частиц, претерпевших превращение, к числу поглощенных веществом квантов света.
Квантовый выход реакции может варьироваться в очень широких пределах: от 10-3 (фотохимическое разложение метилбромида) до 106 (цепная реакция водорода с хлором); в общем случае, чем более долгоживущей является активная частица, тем с большим квантовым выходом протекает фотохимическая реакция.
Важнейшими фотохимическими реакциями являются реакции фотосинтеза, протекающие в растениях с участием хлорофилла:
Процесс фотосинтеза составляют две стадии: световая, связанная с поглощением фотонов, и значительно более медленная темновая, представляющая собой ряд химических превращений, осуществляемых в отсутствие света. Суммарный процесс фотосинтеза заключается в окислении воды до кислорода и восстановлении диоксида углерода до углеводов:
СО2 + Н2О + hν ––> (СН2О) + О2, ΔG° = 477.0 кДж/моль
Протекание данного окислительно-восстановительного процесса (связанного с переносом электронов) возможно благодаря наличию в реакционном центре хлорофилла Сhl донора D и акцептора A электронов; перенос электронов происходит в результате фотовозбуждения молекулы хлорофилла:
DChlA + hν ––> DChl*A ––> DChl+A– ––> D+ChlA–
Возникающие в данном процессе заряженные частицы D+ и A– принимают участие в дальнейших окислительно-восстановительных реакциях темновой стадии фотосинтеза.
2.3 КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
|
[Cu]:СО + Н2 ––> СН3ОН |
[Al2О3]: С2Н5ОН > С2Н4 + Н2О |
|
[Ni]: СО + Н2 ––> СН4 + Н2О |
[Cu]: С2Н5ОН ––> СН3СНО + Н2 |
Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора (рис. 2.8).
Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Действительно, если предположить, что предэкспоненциальные множители в уравнении Аррениуса (II.32) для каталитической и некаталитической реакций близки, то для отношения констант скорости можно записать:
![]() (II.44)
(II.44)
Если ΔEA = –50 кДж/моль, то отношение констант скоростей составит 2,7·106 раз (действительно, на практике такое уменьшение EA увеличивает скорость реакции приблизительно в 105 раз).
Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса (процесса, ΔG (ΔF) которого больше нуля). Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
В зависимости от фазового состояния реагентов и
катализатора различают гомогенный и гетерогенный катализ.
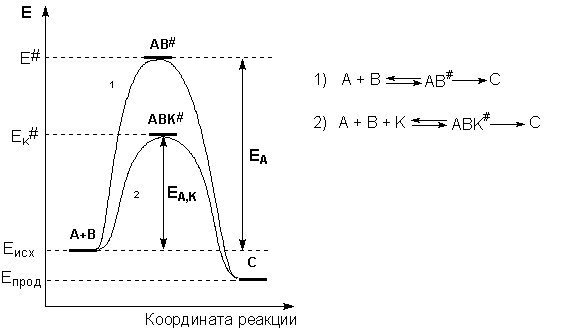
Рис. 2.8 Энергетическая диаграмма химической реакции без катализатора (1) и в присутствии катализатора (2).
2.3.1 Гомогенный катализ.
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль:
СН3СНО ––> СН4 + СО
В присутствии паров йода этот процесс протекает в две стадии:
СН3СНО + I2 ––> СН3I + НI + СО
СН3I + НI ––> СН4 + I2
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
2.3.2Автокатализ.
Автокатализ – процесс
каталитического ускорения химической реакции одним из её продуктов. В качестве
примера можно привести катализируемую ионами водорода реакцию гидролиза сложных
эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием
протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической
реакции состоит в том, что данная реакция протекает с постоянным возрастанием
концентрации катализатора. Поэтому в начальный период реакции скорость е
возрастает, а на последующих стадиях в результате убыли концентрации реагентов
скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической
реакции имеет характерный S-образный вид (рис. 2.9).
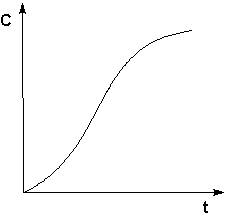
Рис. 2.9
Кинетическая кривая продукта автокаталитической реакции
2.3.3 Гетерогенный катализ.
Гетерогенный катализ каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Например, согласно теории мультиплетов, дегидрирование предельных одноатомных спиртов происходит на дублете, а дегидрирование циклогексана – на секстете (рис. 2.10 – 2.11); теория мультиплетов позволила связать каталитическую активность металлов с величиной их атомного радиуса.
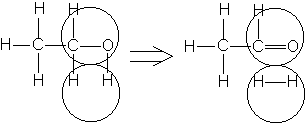 Рис. 2.10 Дегидрирование
спиртов на дублете
Рис. 2.10 Дегидрирование
спиртов на дублете
Рис. 2.11 Дегидрирование циклогексана на секстете
2.3.4 Ферментативный катализ.
Ферментативный катализ каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами. Так, константа скорости реакции разложения перекиси водорода, катализируемой ионами Fе2+, составляет 56 с-1; константа скорости этой же реакции, катализируемой ферментом каталазой, равна 3.5·107, т.е. реакция в присутствии фермента протекает в миллион раз быстрее (энергии активации процессов составляют соответственно 42 и 7.1 кДж/моль). Константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на тринадцать порядков, составляя 7.4·10-7 и 5·106 с-1 (величина энергии активации составляет соответственно 103 и 28 кДж/моль).
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно общепринятым представлениям о механизме ферментативного катализа, субстрат S и фермент F находятся в равновесии с очень быстро образующимся фермент-субстратным комплексом FS, который сравнительно медленно распадается на продукт реакции P с выделением свободного фермента; т.о., стадия распада фермент-субстратного комплекса на продукты реакции является скоростьопределяющей (лимитирующей).
F + S <––> FS ––> F + P
Исследование зависимости скорости ферментативной реакции от концентрации субстрата при неизменной концентрации фермента показали, что с увеличением концентрации субстрата скорость реакции сначала увеличивается, а затем перестает изменяться (рис. 2.12) и зависимость скорости реакции от концентрации субстрата описывается следующим уравнением:
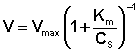 (II.45)
(II.45)
Здесь Кm – константа Михаэлиса, численно равная концентрации субстрата при V = ½Vmax. Константа Михаэлиса служит мерой сродства между субстратом и ферментом: чем меньше Кm, тем больше их способность к образованию фермент-субстратного комплекса.
Характерной особенностью действия ферментов является также высокая чувствительность активности ферментов к внешним условиям рН среды и температуре. Ферменты активны лишь в достаточно узком интервале рН и температуры, причем для ферментов характерно наличие в этом интервале максимума активности при некотором оптимальном значении рН или температуры; по обе стороны от этого значения активность ферментов быстро снижается.
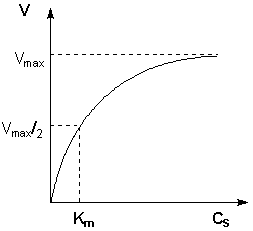
Рис. 2.12 Зависимость скорости ферментативной реакции от концентрации субстрата.
3 ТЕРМОДИНАМИКА РАСТВОРОВ
Существование абсолютно чистых веществ невозможно всякое вещество обязательно содержит примеси, или, иными словами, всякая гомогенная система многокомпонентна. Если имеющиеся в веществе примеси в пределах точности описания системы не оказывают влияния на изучаемые свойства, можно считать систему однокомпонентной; в противном случае гомогенную систему считают раствором.
Раствор гомогенная система, состоящая из двух или более компонентов, состав которой может непрерывно изменяться в некоторых пределах без скачкообразного изменения её свойств.
Раствор может иметь любое агрегатное состояние; соответственно их разделяют на твердые, жидкие и газообразные (последние обычно называют газовыми смесями). Обычно компоненты раствора разделяют на растворитель и растворенное вещество. Как правило, растворителем считают компонент, присутствующий в растворе в преобладающем количестве либо компонент, кристаллизующийся первым при охлаждении раствора; если одним из компонентов раствора является жидкое в чистом виде вещество, а остальными – твердые вещества либо газы, то растворителем считают жидкость. С термодинамической точки зрения это деление компонентов раствора не имеет смысла и носит поэтому условный характер.
Одной из важнейших характеристик раствора является его состав, описываемый с помощью понятия концентрация раствора. Ниже дается определение наиболее распространенных способов выражения концентрации и формулы для пересчета одних концентраций в другие, где индексы А и В относятся соответственно к растворителю и растворенному веществу.
Молярная концентрация С – число молей νВ растворенного вещества в одном литре раствора.
Нормальная концентрация N – число молей эквивалентов растворенного вещества (равное числу молей νВ, умноженному на фактор эквивалентности f) в одном литре раствора.
Моляльная концентрация m – число молей растворенного вещества в одном килограмме растворителя.
Процентная концентрация ω – число граммов растворенного вещества в 100 граммах раствора.
![]() (III.1)
(III.1)
![]() (III.2)
(III.2)
![]() (III.3)
(III.3)
Еще одним способом выражения концентрации является мольная доля X – отношение числа молей данного компонента к общему числу молей всех компонентов в системе.
3.1 ОБРАЗОВАНИЕ РАСТВОРОВ. РАСТВОРИМОСТЬ
Концентрация компонента в растворе может изменяться от нуля до некоторого максимального значения, называемого растворимостью компонента. Растворимость S – концентрация компонента в насыщенном растворе. Насыщенный раствор – раствор, находящийся в равновесии с растворенным веществом. Величина растворимости характеризует равновесие между двумя фазами, поэтому на неё влияют все факторы, смещающие это равновесие (в соответствии с принципом Ле Шателье – Брауна).
Образование раствора является сложным физико-химическим процессом. Процесс растворения всегда сопровождается увеличением энтропии системы; при образовании растворов часто имеет место выделение либо поглощение теплоты. Теория растворов должна объяснять все эти явления. Исторически сложились два подхода к образованию растворов – физическая теория, основы которой были заложены в XIX веке, и химическая, одним из основоположников которой был Д.И. Менделеев. Физическая теория растворов рассматривает процесс растворения как распределение частиц растворенного вещества между частицами растворителя, предполагая отсутствие какого-либо взаимодействия между ними. Единственной движущей силой такого процесса является увеличение энтропии системы ΔS; какие-либо тепловые или объемные эффекты при растворении отсутствуют (ΔН = 0, ΔV = 0; такие растворы принято называть идеальными). Химическая теория рассматривает процесс растворения как образование смеси неустойчивых химических соединений переменного состава, сопровождающееся тепловым эффектом и изменением объема системы (контракцией), что часто приводит к резкому изменению свойств растворенного вещества (так, растворение бесцветного сульфата меди СuSО4 в воде приводит к образованию окрашенного раствора, из которого выделяется не СuSО4, а голубой кристаллогидрат СuSО4·5Н2О). Современная термодинамика растворов основана на синтезе этих двух подходов.
В общем случае при растворении происходит изменение свойств и растворителя, и растворенного вещества, что обусловлено взаимодействием частиц между собой по различным типам взаимодействия: Ван-дер-Ваальсового (во всех случаях), ион-дипольного (в растворах электролитов в полярных растворителях), специфических взаимодействий (образование водородных или донорно-акцепторных связей). Учет всех этих взаимодействий представляет собой очень сложную задачу. Очевидно, что чем больше концентрация раствора, тем интенсивнее взаимодействие частиц, тем сложнее структура раствора. Поэтому количественная теория разработана только для идеальных растворов, к которым можно отнести газовые растворы и растворы неполярных жидкостей, в которых энергия взаимодействия разнородных частиц EA-B близка к энергиям взаимодействия одинаковых частиц EA-A и EB-B. Идеальными можно считать также бесконечно разбавленные растворы, в которых можно пренебречь взаимодействием частиц растворителя и растворенного вещества между собой. Свойства таких растворов зависят только от концентрации растворенного вещества, но не зависят от его природы.
3.1.1 Растворимость газов в газах
Газообразное состояние вещества характеризуется слабым взаимодействием между частицами и большими расстояниями между ними. Поэтому газы смешиваются в любых соотношениях (при очень высоких давлениях, когда плотность газов приближается к плотности жидкостей, может наблюдаться ограниченная растворимость). Газовые смеси описываются законом Дальтона:
Общее давление газовой смеси равно сумме парциальных давлений всех входящих в неё газов.
![]() (III.5)
(III.5)
3.1.2 Растворимость газов в жидкостях
Растворимость газов в жидкостях зависит от ряда факторов: природы газа и жидкости, давления, температуры, концентрации растворенных в жидкости веществ (особенно сильно влияет на растворимость газов концентрация электролитов).
Наибольшее влияние на растворимость газов в жидкостях оказывает природа веществ. Так, в 1 литре воды при t = 18 °С и P = 1 атм. растворяется 0.017 л. азота, 748.8 л. аммиака или 427.8 л. хлороводорода. Аномально высокая растворимость газов в жидкостях обычно обусловливается их специфическим взаимодействием с растворителем – образованием химического соединения (для аммиака) или диссоциацией в растворе на ионы (для хлороводорода). Газы, молекулы которых неполярны, растворяются, как правило, лучше в неполярных жидкостях – и наоборот. Зависимость растворимости газов от давления выражается законом Генри – Дальтона:
Растворимость газа в жидкости прямо пропорциональна его давлению над жидкостью.
![]() (III.7)
(III.7)
Здесь С – концентрация раствора газа в жидкости, k коэффициент пропорциональности, зависящий от природы газа. Закон Генри Дальтона справедлив только для разбавленных растворов при малых давлениях, когда газы можно считать идеальными. Газы, способные к специфическому взаимодействию с растворителем, данному закону не подчиняются.
Растворимость газов в жидкостях существенно зависит от температуры; количественно данная зависимость определяется уравнением Клапейрона Клаузиуса (здесь X – мольная доля газа в растворе, λ – тепловой эффект растворения 1 моля газа в его насыщенном растворе):
![]() (III.8)
(III.8)
Как правило, при растворении газа в жидкости выделяется теплота (λ < 0), поэтому с повышением температуры растворимость уменьшается. Растворимость газов в жидкости сильно зависит от концентрации других растворенных веществ. Зависимость растворимости газов от концентрации электролитов в жидкости выражается формулой Сеченова (X и Xo растворимость газа в чистом растворителе и растворе электролита с концентрацией C):
3.1.3 Взаимная растворимость жидкостей
В зависимости от природы жидкости могут смешиваться в любых соотношениях (в этом случае говорят о неограниченной взаимной растворимости), быть практически нерастворимыми друг в друге либо обладать ограниченной растворимостью. Рассмотрим последний случай на примере системы анилин – вода. Если смешать примерно равные количества воды и анилина, система будет состоять из двух слоев жидкости; верхний слой – раствор анилина в воде, нижний – раствор воды в анилине. Для каждой температуры оба раствора имеют строго определенный равновесный состав, не зависящий от количества каждого из компонентов.
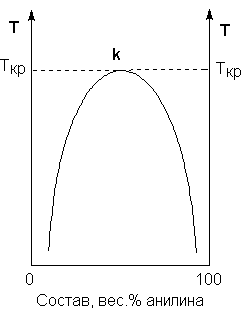
Рис. 3.1 Диаграмма растворимости
системы анилин – вода
Зависимость концентрации растворов от температуры принято изображать графически с помощью диаграммы взаимной растворимости. Эта диаграмма для системы анилин-вода приведена на рис. 3.1. Область под кривой это область расслаивания жидкостей. Повышение температуры приводит к увеличению концентрации каждого из растворов (увеличению взаимной растворимости), и при некоторой температуре, называемой критической температурой расслоения (Ткр на рис. 3.1) взаимная растворимость воды и анилина становится неограниченной. Система анилин – вода относится к т.н. системам с верхней критической температурой расслоения; существуют также и системы, для которых повышение температуры приводит к уменьшению взаимной растворимости компонентов.
3.1.4 Растворимость твердых веществ в жидкостях
Растворимость твердых веществ в жидкостях определяется природой веществ и, как правило, существенно зависит от температуры; сведения о растворимости твердых тел целиком основаны на опытных данных. Качественным обобщением экспериментальных данных по растворимости является принцип "подобное в подобном": полярные растворители хорошо растворяют полярные вещества и плохо – неполярные, и наоборот.
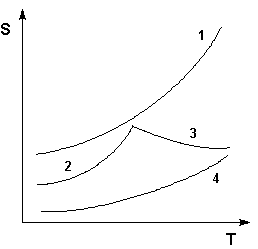
Рис. 3.2 Кривые растворимости
некоторых солей в воде.
1 – КNО3, 2 – Nа2SО4·10Н2О, 3 – Nа2SО4,
4 – Ва(NО3)2
Зависимость растворимости S от температуры обычно изображают графически в
виде кривых растворимости (рис. 3.2). Поскольку теплота растворения твердых
веществ в жидкостях может быть как положительной, так и отрицательной,
растворимость при увеличении температуры может увеличиваться либо уменьшаться
(согласно принципу Ле Шателье – Брауна).
3.2 РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ
3.2.1 Давление насыщенного пара разбавленных растворов
Представим, что в равновесную систему жидкость А пар введено некоторое вещество В. При образовании раствора мольная доля растворителя XА становится меньше единицы; равновесие в соответствии с принципом Ле Шателье – Брауна смещается в сторону конденсации вещества А, т.е. в сторону уменьшения давления насыщенного пара РА. Очевидно, что, чем меньше мольная доля компонента А в растворе, тем меньше парциальное давление его насыщенных паров над раствором. Для некоторых растворов выполняется следующая закономерность, называемая первым законом Рауля:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причем коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
![]() (III.10)
(III.10)
Поскольку сумма мольных долей всех компонентов раствора равна единице, для бинарного раствора, состоящего из компонентов А и В легко получить следующее соотношение, также являющееся формулировкой первого закона Рауля:
![]() (III.11)
(III.11)
Относительное понижение давления пара растворителя над раствором равно мольной доле растворенного вещества и не зависит от природы растворенного вещества.
Растворы, для которых выполняется закон Рауля, называют идеальными растворами. Идеальными при любых концентрациях являются растворы, компоненты которых близки по физическим и химическим свойствам (оптические изомеры, гомологи и т.п.) и образование которых не сопровождается объёмными и тепловыми эффектами. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором. Растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области бесконечно малых концентраций.
3.2.2 Давление пара идеальных и реальных растворов
Если компоненты бинарного (состоящего из двух компонентов) раствора летучи, то пар над раствором будет содержать оба компонента (относительное содержание компонентов в парах будет, как правило, отличаться от содержания их в растворе – пар относительно богаче компонентом, температура кипения которого ниже). Рассмотрим бинарный раствор, состоящий из компонентов А и В, неограниченно растворимых друг в друге. Общее давление пара, согласно первому закону Рауля, равно
![]() (III.12)
(III.12)
Таким образом, для идеальных бинарных растворов
зависимость общего и парциального давления насыщенного пара от состава
раствора, выраженного в мольных долях компонента В, является линейной при любых
концентрациях (рис.3.3). К таким системам относятся, например, системы бензол
толуол, гексан – гептан, смеси изомерных углеводородов и др.
Рис. 3.3 Зависимость парциальных и общего давлений пара идеального раствора от концентрации
Для реальных растворов данные зависимости
являются криволинейными. Если молекулы данного компонента взаимодействуют друг
с другом сильнее, чем с молекулами другого компонента, то истинные парциальные
давления паров над смесью будут больше, чем вычисленные по первому закону Рауля
(положительные отклонения). Если же однородные частицы взаимодействуют
друг с другом слабее, чем разнородные, парциальные давления паров компонентов
будут меньше вычисленных (отрицательные отклонения). Реальные растворы с
положительными отклонениями давления пара образуются из чистых компонентов с
поглощением теплоты (ΔНраств > 0), растворы с отрицательными
отклонениями образуются с выделением теплоты (ΔНраств < 0).
Рис. 3.4 Зависимость парциальных и общего давлений пара идеальных (штриховая линия) и реальных (сплошная линия) бинарных растворов от состава при положительных (слева) и отрицательных (справа) отклонениях от закона Рауля.
3.2.3 Температура кристаллизации разбавленных растворов
Раствор в отличие от чистой жидкости не отвердевает целиком при постоянной температуре; при некоторой температуре, называемой температурой начала кристаллизации, начинают выделяться кристаллы растворителя и по мере кристаллизации температура раствора понижается (поэтому под температурой замерзания раствора всегда понимают именно температуру начала кристаллизации). Замерзание растворов можно охарактеризовать величиной понижения температуры замерзания ΔТзам, равной разности между температурой замерзания чистого растворителя T°зам и температурой начала кристаллизации раствора Tзам:
![]() (III.13)
(III.13)
Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис. 3.5), на которой кривая ОF есть зависимость давления пара над твердым растворителем, а кривые ОА, ВС, DE зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями соответственно. Кристаллы растворителя будут находиться в равновесии с раствором только тогда, когда давление насыщенного пара над кристаллами и над раствором одинаково. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, температура, отвечающая этому условию, всегда будет более низкой, чем температура замерзания чистого растворителя. При этом понижение температуры замерзания раствора ΔTзам не зависит от природы растворенного вещества и определяется лишь соотношением числа частиц растворителя и растворенного вещества.
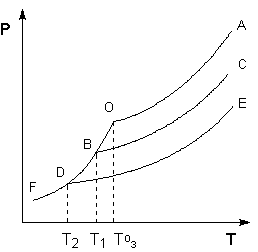
Рис. 3.5 Понижение температуры замерзания
разбавленных растворов
Можно показать, что понижение температуры замерзания раствора ΔTзам прямо пропорционально моляльной концентрации раствора:
![]() (III.14)
(III.14)
Уравнение (III.14) называют вторым законом Рауля. Коэффициент пропорциональности K – криоскопическая постоянная растворителя определяется природой растворителя.
3.2.4 Температура кипения разбавленных растворов
Температура кипения растворов нелетучего вещества всегда выше, чем температура кипения чистого растворителя при том же давлении. Рассмотрим Р – T диаграмму состояния растворителя и растворов различной концентрации (рис.3.5). Любая жидкость – растворитель или раствор – кипит при той температуре, при которой давление насыщенного пара становится равным внешнему давлению. Соответственно температуры, при которых изобара Р = 1 атм. пересечет кривые ОА, ВС и DE, представляющие собой зависимости давления пара над чистым растворителем и растворами с возрастающими концентрациями соответственно, будут температурами кипения этих жидкостей (рис. 3.6).
Повышение температуры кипения растворов нелетучих веществ ΔTк = Tк – T°к пропорционально понижению давления насыщенного пара и, следовательно, прямо пропорционально моляльной концентрации раствора. Коэффициент пропорциональности E есть эбулиоскопическая постоянная растворителя, не зависящая от природы растворенного вещества.
![]() (III.15)
(III.15)
Рис. 3.6 Повышение температуры кипения
разбавленных растворов
Т.о., второй закон Рауля можно в наиболее общем виде сформулировать следующим образом:
Понижение температуры замерзания и повышение температуры кипения разбавленного раствора нелетучего вещества прямо пропорционально моляльной концентрации раствора и не зависит от природы растворенного вещества.
Второй закон Рауля является следствием из первого; данный закон справедлив только для бесконечно разбавленных растворов. Коэффициенты пропорциональности в уравнениях (III.14 – III.15) – эбулиоскопическая и криоскопическая константы – имеют физический смысл соответственно повышения температуры кипения и понижения температуры замерзания растворов с моляльной концентрацией, равной 1 моль/кг. Однако, поскольку такие растворы не являются бесконечно разбавленными, эбулиоскопическая и криоскопическая константы не могут быть непосредственно определены и относятся поэтому к числу т.н. экстраполяционных констант.
3.2.5 Осмотическое давление разбавленных растворов
Если разделить два раствора с различной концентрацией полупроницаемой перегородкой, пропускающей молекулы растворителя, но препятствующей переходу частиц растворённого вещества, будет наблюдаться явление самопроизвольного перехода растворителя через мембрану из менее концентрированного раствора в более концентрированный – осмос. Осмотические свойства раствора количественно характеризуются величиной осмотического давления. Давление, которое необходимо приложить к раствору, чтобы предотвратить перемещение растворителя в раствор через мембрану, разделяющую раствор и чистый растворитель, есть осмотическое давление π. Осмотическое давление идеальных растворов линейно зависит от температуры и молярной концентрации раствора С и может быть рассчитано по уравнению (III.16):
![]() (III.16)
(III.16)
Уравнение (III.16) есть т.н. принцип Вант-Гоффа:
осмотическое давление идеального раствора равно тому давлению, которое оказывало бы растворенное вещество, если бы оно, находясь в газообразном состоянии при той же температуре, занимало бы тот же объем, который занимает раствор.
Осмос играет важнейшую роль в процессах жизнедеятельности животных и растений, поскольку клеточная плазматическая мембрана является полупроницаемой. Осмос обусловливает поднятие воды по стеблю растений, рост клетки и многие другие явления.
Рассмотрим роль осмоса в водном режиме растительной клетки. Осмотическое давление жидкости, контактирующей с клеткой, может быть больше, меньше либо равно осмотическому давлению внутриклеточной жидкости. Соответственно выделяют гипертонические, гипотонические и изотонические растворы.
Если клетка находится в контакте с гипертоническим раствором, вода выходит из неё путём осмоса через плазматическую мембрану. Протопласт (живое содержимое клетки) при этом уменьшается в объёме, сморщивается и в конце концов отстаёт от клеточной стенки. Этот процесс называют плазмолизом. Процесс плазмолиза обычно обратим.
Если клетку поместить в чистую воду или гипотонический раствор, вода путём осмоса поступает в клетку; протопласт при этом увеличивается в объёме и оказывает давление на сравнительно жёсткую клеточную стенку. Этот процесс называется тургором. Тургорное давление препятствует дальнейшему поступлению воды в клетку. Именно тургорное давление поддерживает стебли растений в вертикальном положении, придаёт растениям прочность и устойчивость.
Изотонические растворы не оказывают влияния на водный режим клетки.
У животных клеток нет клеточной стенки, поэтому они более чувствительны к осмотическому давлению жидкости, в которой находятся. Животные клетки имеют систему защиты, основанную на осморегуляции; организм животного стремится поддерживать осмотическое давление всех тканевых жидкостей на постоянном уровне. Например, осмотическое давление крови человека 800 000 Н/м2. Такое же осмотическое давление имеет 0,9 %-ный раствор хлорида натрия. Физиологический раствор, изотоничный крови, широко применяется в медицине.
3.2.6 Понятие активности растворенного вещества
Если концентрация растворенного вещества не превышает 0.1 моль/л, раствор неэлектролита обычно считают разбавленным. В таких растворах взаимодействие между молекулами растворителя существенно преобладает над взаимодействием между молекулами растворителя и растворенного вещества, поэтому последним обычно можно пренебречь. В случае более концентрированных растворов такое приближение неправомерно и для формального учета взаимодействия частиц растворителя и растворенного вещества, а также частиц растворенного вещества между собой, вводится эмпирическая величина, заменяющая концентрацию – активность (эффективная концентрация) а, связанная с концентрацией через коэффициент активности f, который является мерой отклонения свойств реального раствора от идеального:
![]() (III.17)
(III.17)
Как правило, коэффициент активности меньше единицы (при малых концентрациях считают f = 1 и а = С). Необходимо отметить, что активность компонента не прямо пропорциональна его концентрации – коэффициент активности уменьшается с увеличением концентрации.
3.3 РАСТВОРЫ ЭЛЕКТРОЛИТОВ
3.3.1 Теория электролитической диссоциации
Законы Рауля и принцип Вант-Гоффа не выполняются для растворов (даже бесконечно разбавленных), которые проводят электрический ток растворов электролитов. Обобщая экспериментальные данные, Я.Г. Вант-Гофф пришел к выводу, что растворы электролитов всегда ведут себя так, будто они содержат больше частиц растворенного вещества, чем следует из аналитической концентрации: повышение температуры кипения, понижение температуры замерзания, осмотическое давление для них всегда больше, чем вычисленные. Для учета этих отклонений Вант-Гофф внес в уравнение (III.16) для растворов электролитов поправку – изотонический коэффициент i:
![]() (III.18)
(III.18)
Аналогичная поправка вносится в законы Рауля, и изотонический коэффициент определяется следующим образом:
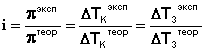 (III.19)
(III.19)
Изотонический коэффициент для растворов электролитов всегда больше единицы, причем с разбавлением раствора i возрастает до некоторого целочисленного значения.
Для объяснения особенностей свойств растворов электролитов С. Аррениус предложил теорию электролитической диссоциации, основывающуюся на следующих постулатах:
1. Электролиты в растворах распадаются на ионы диссоциируют;
2. Диссоциация является обратимым равновесным процессом;
3. Силы взаимодействия ионов с молекулами растворителя и друг с другом малы (т.е. растворы являются идеальными).
Диссоциация электролитов в растворе происходит под действием полярных молекул растворителя; наличие ионов в растворе предопределяет его электропроводность. Для оценки полноты диссоциации в теории электролитической диссоциации вводится понятие степень диссоциации α, которая равна отношению числа молекул n, распавшихся на ионы, к общему числу молекул N:
![]() (III.20)
(III.20)
Величина степени диссоциации зависит от природы растворителя и растворенного вещества, концентрации раствора и температуры. По величине степени диссоциации электролиты подразделяются на три группы: сильные (α ≥ 0.7), средней силы (0.3 < α < 0.7) и слабые (α ≤ 0.3). К сильным электролитам относятся почти все соли (кроме Рb(СН3СОО)2, НgСl2, СdСl2), большинство неорганических кислот и щелочей; к слабым – все органические кислоты, вода, NН4ОН, Н2S и т.д. Электролитами средней силы являются некоторые неорганические кислоты: НF, НСN, Н3PO4.
3.3.2 Слабые электролиты. Константа диссоциации
Процесс диссоциации слабых электролитов является обратимым и в системе существует динамическое равновесие, которое может быть описано константой равновесия, выраженной через концентрации образующихся ионов и непродиссоциировавших молекул, называемой константой диссоциации. Для некоторого электролита, распадающегося в растворе на ионы в соответствии с уравнением:
АaВb <––> aАx- + bВy+
константа диссоциации выразится следующим соотношением:
![]() (III.21)
(III.21)
Для бинарного (распадающегося на два иона) электролита выражение (III.21) можно переписать в виде (III.21a):
![]() (III.21a)
(III.21a)
Поскольку концентрация каждого иона для бинарного электролита равна произведению степени диссоциации α на общую концентрацию электролита С, выражение (III.21a) в этом случае можно переписать следующим образом:
![]() (III.22)
(III.22)
Для разбавленных растворов можно считать, что (1 α) = 1. Тогда получаем:
![]() (III.23)
(III.23) ![]() (III.24)
(III.24)
Т.о., степень диссоциации слабого электролита обратно пропорциональна концентрации и прямо пропорциональна разбавлению раствора; выражение (III.24) называют законом разбавления Оствальда. Степень диссоциации слабого электролита можно связать с изотоническим коэффициентом. Будем считать, что из N молекул электролита продиссоциировало n молекул, образовав νn ионов (ν – число ионов, на которое диссоциирует молекула). Поскольку изотонический коэффициент показывает, во сколько раз общее число молекул и ионов в растворе больше числа молекул до диссоциации, получаем:
![]() (III.25)
(III.25)
![]() (III.26)
(III.26)
Соотношение (III.26) дает возможность, экспериментально определив изотонический коэффициент раствора, рассчитать степень диссоциации слабого электролита.
3.3.3 Сильные электролиты
Предположение Аррениуса о том, что в растворе сильного электролита также существует динамическое равновесие между молекулами и ионами, как и у слабых электролитов, оказалось ошибочным. Экспериментальные исследования показали, что, во-первых, величина константы диссоциации сильного электролита зависит от концентрации (т.е. к растворам сильных электролитов неприменим закон действующих масс) и, во-вторых, никакими методами не удалось обнаружить в растворах сильных электролитов непродиссоциировавшие молекулы. Это позволило сделать вывод, что сильные электролиты в растворах любых концентраций полностью диссоциируют на ионы и, следовательно, закономерности, полученные для слабых электролитов, не могут применяться к сильным электролитам без соответствующих поправок.
Качественная теория сильных электролитов была разработана П. Дебаем и Г. Хюккелем (1923). Для сильных электролитов, полностью диссоциирующих на ионы, даже при малых концентрациях растворов энергия электростатического взаимодействия между ионами достаточно велика, и пренебречь этим взаимодействием нельзя. Взаимодействие противоположно и одноименно заряженных ионов (соответственно притяжение и отталкивание) приводит к тому, что вблизи каждого иона находятся преимущественно ионы с противоположным зарядом, образующие т.н. ионную атмосферу. Радиус ионной атмосферы сравнительно велик, поэтому ионные атмосферы соседних ионов пересекаются; кроме того, каждый ион окружен дипольными молекулами растворителя сольватной оболочкой. Т.о., в растворе сильного электролита возникает подобие пространственной структуры, что ограничивает свободу перемещения ионов и приводит к изменению свойств раствора в том же направлении, как действовало бы уменьшение степени диссоциации. Поэтому, определяя степень диссоциации раствора сильного электролита, получают т.н. кажущуюся степень диссоциации, т.е. величину α с поправкой на межионное взаимодействие. Чем выше концентрация раствора, тем сильнее взаимодействие ионов, тем меньше и кажущаяся степень диссоциации сильного электролита.
Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятий активности электролита аэ и активностей катионов и анионов а+ и а- соответственно, которые равны произведению коэффициента активности на концентрацию:
![]() ;
; ![]() ;
; ![]() (III.27)
(III.27)
Для бинарного электролита средняя активность электролита связана с активностями ионов соотношением (III.28); подобным же образом связан средний коэффициент активности с ионными:
![]() (III.28)
(III.28)
![]() (III.29)
(III.29)
Дебаем и Хюккелем был разработан метод расчета среднего коэффициента активности сильного электролита. Для бинарного электролита уравнение имеет следующий вид:
![]() (III.30)
(III.30)
Здесь z – заряд иона, для которого рассчитывается коэффициент активности, I – т.н. ионная сила раствора: некоторый параметр, который одновременно учитывает молярную концентрацию и заряд всех имеющихся в растворе ионов. Ионная сила раствора равна полусумме концентраций всех ионов, умноженных на квадрат их заряда:
![]() (III.31)
(III.31)
Теория Дебая – Хюккеля применима только при концентрациях, не превышающих 0.05 моль/л. Для более концентрированных растворов сильных электролитов количественной теории не существует.
3.4 ЭЛЕКТРОПРОВОДНОСТЬ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
3.4.1 Удельная электропроводность растворов электролитов
Электрический ток есть упорядоченное перемещение заряженных частиц. Растворы электролитов обладают ионной проводимостью (являются т.н. проводниками второго рода), т.е. электропроводность растворов электролитов обусловлена перемещением ионов в электрическом поле (в отличие от электронной проводимости проводников первого рода).
Величина преимущественного передвижения иона в направлении одного из электродов при прохождении тока через раствор отнесённая к градиенту потенциала 1 В/см, есть абсолютная скорость движения иона. Абсолютные скорости движения ионов имеют величины порядка 0,0005 – 0,003 см2/(В·с). Абсолютные скорости движения катионов U+ и анионов U– различаются; это приводит к тому, что ионы разных знаков переносят разные количества электричества.
Всякий проводник, по которому течет ток, представляет для него определенное сопротивление R, которое, согласно закону Ома, прямо пропорционально длине проводника l и обратно пропорционально площади сечения S; коэффициентом пропорциональности является удельное сопротивление материала ρ – сопротивление проводника, имеющего длину 1 см и сечение 1 см2:
![]() , Ом (III.32)
, Ом (III.32)
В качестве количественной меры способности раствора электролита проводить электрический ток используют обычно удельную электропроводность κ (каппа) – величину, обратную удельному сопротивлению (т.е. величину, обратную сопротивлению столба раствора между электродами площадью 1 см2, находящимися на расстоянии 1 см):
![]() , Ом-1см-1
(III.33)
, Ом-1см-1
(III.33)
Величина удельной электропроводности электролита
зависит от ряда факторов: природы электролита, температуры, концентрации
раствора. Удельная электропроводность растворов электролитов (в отличие от
электропроводности проводников первого рода) с увеличением температуры
возрастает, что вызвано увеличением скорости движения ионов за счет понижения
вязкости раствора и уменьшения сольватированности ионов. Зависимость удельной
электропроводности от концентрации раствора представлена на рис. 3.7.
Рис. 3.7 Зависимость удельной
электропроводности электролитов от концентрации
(1 – H2SO4, 2 – KOH, 3 – CH3COOH)
Как видно из рисунка, с увеличением концентрации удельная электропроводность растворов сначала возрастает, достигая некоторого максимального значения, затем начинает уменьшаться. Эта зависимость очень чётко выражена для сильных электролитов и значительно хуже для слабых. Наличие максимума на кривых объясняется тем, что в разбавленных растворах сильных электролитов скорость движения ионов мало зависит от концентрации, и κ сначала растет почти прямо пропорционально числу ионов; с ростом концентрации усиливается взаимодействие ионов, что уменьшает скорость их движения. Для слабых электролитов наличие максимума на кривой обусловлено тем, что с ростом концентрации уменьшается степень диссоциации, и при достижении определенной концентрации число ионов в растворе начинает увеличиваться медленнее, чем концентрация. Для учета влияния на электрическую проводимость растворов электролитов их концентрации и взаимодействия между ионами введено понятие молярной электропроводности раствора.
3.4.2 Молярная электропроводность растворов электролитов
Молярная электропроводность раствора λ есть величина, обратная сопротивлению раствора, содержащего 1 моль растворенного вещества и помещенного между электродами, расположенными на расстоянии 1 см друг от друга. С удельной электропроводностью κ и молярной концентрацией раствора С молярная электропроводность связана следующим соотношением:
![]() , Ом-1см2моль-1
(III.34)
, Ом-1см2моль-1
(III.34)
Молярная электропроводность как сильных, так и слабых электролитов увеличивается с уменьшением концентрации (т.е. увеличением разведения раствора V = 1/С), достигая некоторого предельного значения λo, называемого молярной электропроводностью при бесконечном разведении (рис. 3.8 3.9).
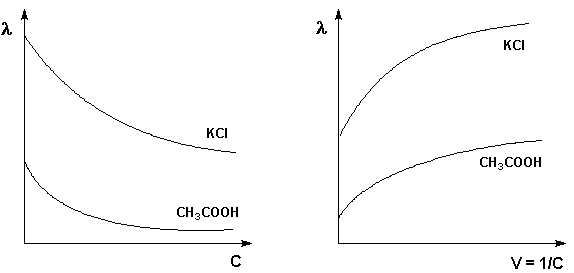
Рис. 3.8 Зависимость молярной Рис. 3.9 Зависимость молярной электропроводности от концентрации. электропроводности от разведения
Для слабого электролита такая зависимость молярной электропроводности от концентрации обусловлена в основном увеличением степени диссоциации с разбавлением раствора. В случае сильного электролита с уменьшением концентрации ослабляется взаимодействие ионов между собой, что увеличивает скорость их движения и, следовательно, молярную электропроводность раствора. Последнюю связывает с абсолютными скоростями движения катионов и анионов U+ и U– уравнение Аррениуса (III.35):
![]() (III.35)
(III.35)
Ф. Кольрауш показал, что в молярную электропроводность бесконечно разбавленных растворов электролитов каждый из ионов вносит свой независимый вклад, и λo является суммой молярных электропроводностей катиона и аниона λ+ и λ– (т.н. подвижностей ионов), и сформулировал закон независимости движения ионов:
Молярная электропроводность при бесконечном разведении равна сумме электролитических подвижностей катиона и аниона данного электролита.
![]() (III.36)
(III.36)
Подставив в это выражение уравнение Аррениуса (III.35) и приняв, что при бесконечном разведении степень диссоциации α равна единице, получим:
![]() (III.37)
(III.37)
Отсюда
![]() ;
; ![]() (III.38)
(III.38)
Электролитическая подвижность является важнейшей характеристикой иона, отражающей его участие в электропроводности раствора.
3.5 ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ
3.5.1 Электрические потенциалы на фазовых границах
При соприкосновении проводника первого рода (электрода) с полярным растворителем (водой) либо раствором электролита на границе электрод – жидкость возникает т.н. двойной электрический слой (ДЭС). В качестве примера рассмотрим медный электрод, погруженный в воду либо в раствор сульфата меди.
При погружении медного электрода в воду часть ионов меди, находящихся в узлах кристаллической решетки, в результате взаимодействия с диполями воды будет переходить в раствор. Возникающий при этом на электроде отрицательный заряд будет удерживать перешедшие в раствор ионы в приэлектродном пространстве – образуется двойной электрический слой (рис. 3.10а; о моделях строения ДЭС смотрите п. 4.2.4). Отрицательный заряд на электроде будет препятствовать дальнейшему переходу ионов меди в раствор, и через некоторое время установится динамическое равновесие, которое можно однозначно охарактеризовать потенциалом электрического поля ДЭС Φ, зависящего от заряда на электроде, или некоторой равновесной концентрацией ионов в приэлектродном слое Сo. При погружении медного электрода в раствор СuSО4, содержащий ионы меди в концентрации С возможны три случая:
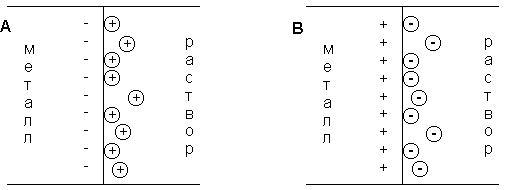
Рис. 3.10 Схема двойного электрического слоя на границе электрод-раствор
1. С < Сo. Поскольку концентрация ионов меди в поверхностном слое меньше равновесной, начнется переход ионов из электрода в раствор; электрод заряжается отрицательно, в поверхностном слое раствора катионов будет больше, чем анионов (рис. 3.9а).
2. С > Сo. Поскольку концентрация ионов меди в поверхностном слое больше равновесной, начнется переход ионов из раствора в электрод; на электроде возникает положительный заряд и в поверхностном слое преобладают анионы SО42- (рис. 3.9b).
3. С = Сo. Поскольку концентрация ионов меди в поверхностном слое равна равновесной (такие растворы называют нулевыми), заряд на электроде не возникает, двойной электрический слой не образуется.
3.5.2 Гальванический элемент. ЭДС гальванического элемента
Рассмотрим простейший гальванический элемент Даниэля-Якоби, состоящий из двух полуэлементов – цинковой и медной пластин, помещенных в растворы сульфатов цинка и меди соответственно, которые соединены между собой посредством электролитического ключа – например, полоски бумаги, смоченной раствором какого-либо электролита. Схематически данный элемент изображается следующим образом:
Zn / Zn2+ // Cu2+ / Cu
На поверхности каждого из электродов имеет место динамическое равновесие перехода ионов металла из электрода в раствор и обратно, характеризуемое потенциалом ДЭС (зарядом на электроде q). Если соединить медный и цинковый электроды металлическим проводником, немедленно произойдет перераспределение зарядов – электроны начнут перемещаться с электрода с более отрицательным зарядом (в нашем случае – цинкового) на электрод с более положительным зарядом (медный), т.е. в проводнике возникнет электрический ток. Изменение величины заряда каждого из электродов нарушает равновесие – на цинковом электроде начнется процесс перехода ионов из электрода в раствор (окисление металла), на медном – из раствора в электрод (восстановление металла); при этом протекание процесса на одном электроде обусловливает одновременное протекание противоположного процесса на другом:
Zno ––> Zn2+ + 2е-
Сu2+ + 2е- > Сuo
Электрод, на котором при работе гальванического элемента протекает процесс окисления, называется анодом, электрод, на котором идет процесс восстановления – катодом. При схематическом изображении гальванических элементов слева записывают анод, справа – катод (стандартный водородный электрод всегда записывают слева). Суммарный окислительно-восстановительный процесс, происходящий в гальваническом элементе, выражается следующим уравнением:
Сu2+ + Zno ––> Сuo + Zn2+
Т.о., гальванический элемент можно определить как прибор для преобразования химической энергии окислительно-восстановительной реакции в электрическую за счет пространственного разделения процессов окисления и восстановления. Работа, которую может совершить электрический ток, вырабатываемый гальваническим элементом, определяется разностью электрических потенциалов между электродами (называемой обычно просто разностью потенциалов) ΔΦ и количеством прошедшего по цепи электричества q:
![]() (III.39)
(III.39)
Работа тока гальванического элемента (и, следовательно, разность потенциалов), будет максимальна при его обратимой работе, когда процессы на электродах протекают бесконечно медленно и сила тока в цепи бесконечно мала. Максимальная разность потенциалов, возникающая при обратимой работе гальванического элемента, есть электродвижущая сила (ЭДС) гальванического элемента.
3.5.3 Электродный потенциал. Уравнение Нернста
ЭДС гальванического элемента E удобно представлять в виде разности некоторых величин, характеризующих каждый из электродов электродных потенциалов; однако для точного определения этих величин необходима точка отсчета – точно известный электродный потенциал какого-либо электрода. Электродным потенциалом электрода εэ называется ЭДС элемента, составленного из данного электрода и стандартного водородного электрода (см. ниже), электродный потенциал которого принят равным нулю. При этом знак электродного потенциала считают положительным, если в таком гальваническом элементе испытуемый электрод является катодом, и отрицательным, если испытуемый электрод является анодом. Необходимо отметить, что иногда электродный потенциал определяют как "разность потенциалов на границе электрод – раствор", т.е. считают его тождественным потенциалу ДЭС, что не вполне правильно (хотя эти величины взаимосвязаны).
Величина электродного потенциала металлического электрода зависит от температуры и активности (концентрации) иона металла в растворе, в который опущен электрод; математически эта зависимость выражается уравнением Нернста (здесь F – постоянная Фарадея, z – заряд иона):
![]() (III.40)
(III.40)
В уравнении Нернста ε° – стандартный электродный потенциал, равный потенциалу электрода при активности иона металла, равной 1 моль/л. Стандартные электродные потенциалы электродов в водных растворах составляют ряд напряжений. Величина ε° есть мера способности окисленной формы элемента или иона принимать электроны, т.е. восстанавливаться. Иногда различием между концентрацией и активностью иона в растворе пренебрегают, и в уравнении Нернста под знаком логарифма фигурирует концентрация ионов в растворе. Величина электродного потенциала определяет направление процесса, протекающего на электроде при работе гальванического элемента. На полуэлементе, электродный потенциал которого имеет большее (иногда говорят – более положительное) значение, будет протекать процесс восстановления, т.е. данный электрод будет являться катодом.
Рассмотрим расчёт ЭДС элемента Даниэля-Якоби с помощью уравнения Нернста. ЭДС всегда является положительной величиной и равна разности электродных потенциалов катода и анода:
![]() (III.41)
(III.41)
![]() (III.42)
(III.42)
![]() (III.43)
(III.43)
![]() (III.44)
(III.44)
![]() (III.45)
(III.45)
Как видно из уравнения (III.45), ЭДС элемента Даниэля-Якоби зависит от концентрации (точнее говоря, активности) ионов меди и цинка; при их равных концентрациях ЭДС элемента будет равна разности стандартных электродных потенциалов:
![]() (III.46)
(III.46)
Анализируя уравнение (III.45), можно определить предел необратимой работы гальванического элемента. Поскольку на аноде идет процесс окисления цинка, концентрация ионов цинка при необратимой работе гальванического элемента постоянно увеличивается; концентрация ионов меди, напротив, уменьшается. Отношение концентраций ионов меди и цинка постоянно уменьшается и логарифм этого отношения при [Сu2+] < [Zn2+] становится отрицательным. Т.о., разность потенциалов при необратимой работе гальванического элемента непрерывно уменьшается; при E = 0 (т.е. εк = εа) гальванический элемент не может совершать работу (необратимая работа гальванического элемента может прекратиться также и в результате полного растворения цинкового анода).
Уравнение (III.45) объясняет также и работоспособность т.н. концентрационных цепей – гальванических элементов, состоящих из двух одинаковых металлических электродов, опущенных в растворы соли этого металла с различными активностями а1 > а2. Катодом в этом случае будет являться электрод с большей концентрацией, т.к. стандартные электродные потенциалы обоих электродов равны; для ЭДС концентрационного гальванического элемента получаем:
![]() (III.47)
(III.47)
Единственным результатом работы концентрационного элемента является перенос ионов металла из более концентрированного раствора в менее концентрированный. Т.о., работа электрического тока в концентрационном гальваническом элементе – это работа диффузионного процесса, который проводится обратимо в результате пространственного разделения его на два противоположных по направлению обратимых электродных процесса.
3.5.4 Классификация электродов
По типу электродной реакции все электроды можно разделить на две группы (в отдельную группу выделяются окислительно-восстановительные электроды, которые будут рассмотрены особо в разделе 3.5.5).
Электроды первого рода
К электродам первого рода относятся электроды, состоящие из металлической пластинки, погруженной в раствор соли того же металла. При обратимой работе элемента, в который включен электрод, на металлической пластинке идет процесс перехода катионов из металла в раствор либо из раствора в металл. Т.о., электроды первого рода обратимы по катиону и их потенциал связан уравнением Нернста (III.40) с концентрацией катиона (к электродам первого рода относят также и водородный электрод).
![]() (III.40)
(III.40)
Электроды второго рода
Электродами второго рода являются электроды, в которых металл покрыт малорастворимой солью этого металла и находится в растворе, содержащем другую растворимую соль с тем же анионом. Электроды этого типа обратимы относительно аниона и зависимость их электродного потенциала от температуры и концентрации аниона может быть записана в следующем виде:
![]() (III.48)
(III.48)
Электроды сравнения
Для определения электродного потенциала элемента необходимо измерить ЭДС гальванического элемента, составленного из испытуемого электрода и электрода с точно известным потенциалом – электрода сравнения. В качестве примеров рассмотрим водородный, каломельный и хлорсеребряный электроды.
Водородный электрод представляет собой платиновую пластинку, омываемую газообразным водородом, погруженную в раствор, содержащий ионы водорода. Адсорбируемый платиной водород находится в равновесии с газообразным водородом; схематически электрод изображают следующим образом:
Рt, Н2 / Н+
Электрохимическое равновесие на электроде можно рассматривать в следующем виде:
2Н+ + 2е- > Н2
Потенциал водородного электрода зависит от активности ионов Н+ в растворе и давления водорода; потенциал стандартного водородного электрода (с активностью ионов Н+ 1 моль/л и давлением водорода 101.3 кПа) принят равным нулю. Поэтому для электродного потенциала нестандартного водородного электрода можно записать:
![]() (III.49)
(III.49)
Каломельный электрод. Работа с водородным электродом довольно неудобна, поэтому в качестве электрода сравнения часто используется более простой в обращении каломельный электрод, величина электродного потенциала которого относительно стандартного водородного электрода точно известна и зависит только от температуры. Каломельный электрод состоит из ртутного электрода, помещенного в раствор КСl определенной концентрации и насыщенный каломелью Hg2Сl2:
Нg / Нg2Сl2, КСl
Каломельный электрод обратим относительно анионов хлора и уравнение Нернста для него имеет вид:
![]() (III.50)
(III.50)
Хлорсеребряный электрод. В качестве электрода сравнения используют также другой электрод второго рода хлорсеребряный, который также обратим относительно анионов хлора:
Аg / АgСl, КСl
Величина потенциала хлорсеребряного электрода зависит от активности ионов хлора; данная зависимость имеет следующий вид:
![]() (III.51)
(III.51)
Чаще всего в качестве электрода сравнения используется насыщенный хлорсеребряный электрод, потенциал которого зависит только от температуры.
Индикаторные электроды.
Электроды, обратимые относительно иона водорода, используются на практике для определения активности этих ионов в растворе (и, следовательно, рН раствора) потенциометрическим методом, основанном на определении потенциала электрода в растворе с неизвестным рН и последующим расчетом рН по уравнению Нернста. В качестве индикаторного электрода может использоваться и водородный электрод, однако работа с ним неудобна и на практике чаще применяются хингидронный и стеклянный электроды.
Хингидронный электрод, относящийся к классу окислительно-восстановительных электродов (см. ниже), представляет собой платиновую проволоку, опущенную в сосуд с исследуемым раствором, в который предварительно помещают избыточное количество хингидрона С6Н4О2·С6Н4(ОН)2 соединения хинона С6Н4О2 и гидрохинона С6Н4(ОН)2, способных к взаимопревращению в равновесном окислительно-восстановительном процессе, в котором участвуют ионы водорода:
С6Н4О2 + 2Н+ + 2е- ––> С6Н4(ОН)2
Хингидронный электрод является т.н. окислительно-восстановительным электродом (см. разд. 3.5.5); зависимость его потенциала от активности ионов водорода имеет следующий вид:
![]() (III.52)
(III.52)
Стеклянный электрод, являющийся наиболее распространенным индикаторным электродом, относится к т.н. ионоселективным или мембранным электродам. В основе работы таких электродов лежат ионообменные реакции, протекающие на границах мембран с растворами электролитов; ионоселективные электроды могут быть обратимы как по катиону, так и по аниону.
Принцип действия мембранного электрода заключается в следующем. Мембрана, селективная по отношению к некоторому иону (т.е. способная обмениваться этим ионом с раствором), разделяет два раствора с различной активностью этого иона. Разность потенциалов, устанавливающаяся между двумя сторонами мембраны, измеряется с помощью двух электродов. При соответствующем составе и строении мембраны её потенциал зависит только от активности иона, по отношению к которому мембрана селективна, по обе стороны мембраны.
Наиболее часто употребляется стеклянный электрод в виде трубки, оканчивающейся тонкостенным стеклянным шариком. Шарик заполняется раствором НСl с определенной активностью ионов водорода; в раствор погружен вспомогательный электрод (обычно хлорсеребряный). Потенциал стеклянного электрода с водородной функцией (т.е. обратимого по отношению к иону Н+) выражается уравнением
![]() (III.53)
(III.53)
Необходимо отметить, что стандартный потенциал ε°ст для каждого электрода имеет свою величину, которая со временем изменяется; поэтому стеклянный электрод перед каждым измерением рН калибруется по стандартным буферным растворам с точно известным рН.
3.5.5 Окислительно-восстановительные электроды
В отличие от описанных электродных процессов в случае окислительно-восстановительных электродов процессы получения и отдачи электронов атомами или ионами происходят не на поверхности электрода, а только в растворе электролита. Если опустить платиновый (или другой инертный) электрод в раствор, содержащий двух- и трехзарядные ионы железа и соединить этот электрод проводником с другим электродом, то возможно либо восстановление ионов Fe3+ до Fe2+ за счет электронов, полученных от платины, либо окисление ионов Fe2+ до Fe3+ с передачей электронов платине. Сама платина в электродном процессе не участвуют, являясь лишь переносчиком электронов. Такой электрод, состоящий из инертного проводника первого рода, помещенного в раствор электролита, содержащего один элемент в различных степенях окисления, называется окислительно-восстановительным или редокс-электродом. Потенциал окислительно-восстановительного электрода также определяют относительно стандартного водородного электрода:
Pt, H2 / 2H+ // Fe3+, Fe2+ / Pt
Зависимость потенциала редокс-электрода εRO от концентрации (активности) окисленной [Ox] и восстановленной форм [Red] для окислительно-восстановительной реакции, в которой не участвуют никакие другие частицы, кроме окислителя и восстановителя, имеет следующий вид (здесь n – число электронов, участвующих в элементарном акте окислительно-восстановительной реакции):
![]() (III.54)
(III.54)
Из данного выражения следует уравнение для потенциала металлического электрода (III.40), т.к. активность атомов металла (восстановленной формы) в материале электрода равна единице.
В случае более сложных систем в выражении для окислительно-восстановительного потенциала фигурируют концентрации всех участвующих в реакции соединений, т.е. под окисленной формой следует понимать все соединения в левой части уравнения реакции
Ох + ne- ––> Red,
а под восстановленной – все соединения в правой части уравнения. Так, для окислительно-восстановительных реакций, протекающих с участием ионов водорода
Ох + ne- + mH+ > Red,
уравнение Нернста будет записываться следующим образом:
![]() (III.55)
(III.55)
При составлении гальванических элементов с участием редокс-электрода электродная реакции на последнем в зависимости от природы второго электрода может быть либо окислительной, либо восстановительной. Например, если составить гальванический элемент из электрода Pt / Fe3+, Fe2+ и второго электрода, имеющего более положительный электродный потенциал, то при работе элемента редокс-электрод будет являться анодом, т.е. на нем будет протекать процесс окисления:
Fe2+ ––> Fe3+ + e-
Если потенциал второго электрода будет меньше, чем потенциал электрода Pt / Fe3+, Fe2+, то на последнем будет протекать реакция восстановления и он будет являться катодом:
Fe3+ + e- > Fe2+
Знание величин электродных потенциалов позволяет определить возможность и направление самопроизвольного протекания любой окислительно-восстановительной реакции при одновременном наличии в растворе двух или более окислительно-восстановительных пар. Восстановленная форма любого элемента или иона будет восстанавливать окисленную форму другого элемента или иона, имеющего более положительный электродный потенциал.
4.1 ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
4.1.1 Поверхностная энергия. Адсорбция
До сих пор свойства гетерогенных систем описывались с помощью параметров и функций состояния, характеризующих каждую из фаз в целом. Однако свойства участка фазы, примыкающего к её поверхности, отличаются от свойств фазы в объеме: фактически частицы, находящиеся на поверхности каждой фазы, образуют особую поверхностную фазу, свойства которой существенно отличаются от свойств внутренних областей фазы. Частицы, расположенные на поверхности, находятся в другом окружении по сравнению с частицами, находящимися в объеме фазы, т.е. взаимодействуют как с однородными частицами, так и с частицами другого рода. Следствием этого является то, что средняя энергия gs частицы, находящейся на поверхности раздела фаз, отличается от средней энергии такой же частицы в объеме фазы gv (причем энергия частицы на поверхности может быть как больше, так и меньше энергии частицы в объеме). Поэтому важнейшей характеристикой поверхностной фазы является поверхностная энергия Gs – разность средней энергии частицы, находящейся на поверхности, и частицы, находящейся в объеме фазы, умноженная на число частиц на поверхности N:
![]() (IV.1)
(IV.1)
![]() (IV.2)
(IV.2)
Очевидно, что общая величина поверхностной энергии фазы будет определяться величиной её поверхности S. Поэтому для характеристики поверхности раздела, отделяющей данную фазу от другой, вводится понятие поверхностное натяжение σ – отношение поверхностной энергии к площади поверхности раздела фаз; величина поверхностного натяжения зависит только от природы обеих фаз. Как и поверхностная энергия фазы, поверхностное натяжение может иметь как положительное, так и отрицательное значение. Поверхностное натяжение положительно, если находящиеся на поверхности частицы взаимодействуют с частицами этой же фазы сильнее, чем с частицами другой фазы (и, следовательно, gs > gv). Согласно принципу минимума свободной энергии, любая фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию; поэтому в случае положительного поверхностного натяжения (σ > 0) фаза стремится уменьшить свою поверхность. В случае если σ < 0, поверхностная энергия фазы будет уменьшаться при увеличении площади поверхности.
Влияние поверхностного слоя фазы на её общие свойства определяется долей частиц, находящихся на поверхности, от общего числа составляющих данную фазу частиц, т.е. величиной удельной поверхности фазы S/V (поверхности, приходящейся на единицу объема). Свободную энергию фазы G можно представить как сумму поверхностной Gs и объемной Gv энергий, пропорциональных соответственно площади поверхности и объему фазы:
![]() (IV.3)
(IV.3)
Разделив это выражение на объем фазы, получаем:
![]() (IV.4)
(IV.4)
Из уравнения (IV.4) следует, что при одном и том же количестве фазы (т.е. неизменном объеме) вклад поверхностной энергии в общую энергию фазы возрастает с увеличением удельной поверхности или, иначе говоря, степени дисперсности (раздробленности) фазы. В случае, когда степень дисперсности фазы невелика (удельная поверхность незначительна), вкладом поверхностной энергии в полную энергию фазы обычно пренебрегают. Вклад поверхностного слоя в свойства фазы и системы в целом учитывают при изучении дисперсных систем гетерогенных систем, одна из фаз которой является сплошной (дисперсионная среда), а другая – раздробленной (дисперсная фаза).
На границе конденсированной (т.е. твердой или жидкой) фазы с газом поверхностное натяжение всегда положительно, поскольку частицы конденсированной фазы взаимодействуют друг с другом сильнее, чем с молекулами газа. Согласно принципу минимума свободной энергии, конденсированная фаза будет стремиться самопроизвольно уменьшить свою поверхностную энергию. Это может быть результатом либо уменьшения площади поверхности фазы (именно поэтому капля жидкости в невесомости принимает форму сферы), либо уменьшения поверхностного натяжения при появлении на поверхности раздела фаз новых частиц – молекул газа либо растворенного вещества. Процесс самопроизвольного изменения концентрации какого-либо вещества у поверхности раздела двух фаз называется адсорбцией. Адсорбентом называется вещество, на поверхности которого происходит изменение концентрации другого вещества – адсорбата.
4.1.2 Адсорбция на границе раствор пар
В жидких растворах поверхностное натяжение σ является функцией от концентрации растворенного вещества. На рис. 4.1 представлены три возможных зависимости поверхностного натяжения от концентрации раствора (т.н. изотермы поверхностного натяжения). Вещества, добавление которых к растворителю уменьшает поверхностное натяжение, называют поверхностно-активными (ПАВ), вещества, добавление которых увеличивает или не изменяет поверхностное натяжение – поверхностно-инактивными (ПИАВ).
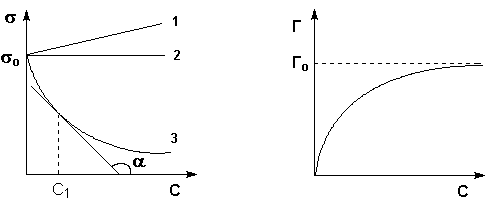
Рис. 4.1 Изотермы поверхностного Рис. 4.2 Изотерма адсорбции натяжения растворов ПАВ (1, 2) и ПАВ на границе раствор – пар ПИАВ (3)
Уменьшение поверхностного натяжения и, следовательно, поверхностной энергии происходит в результате адсорбции ПАВ на поверхности раздела жидкость – пар, т.е. того, что концентрация поверхностно-активного вещества в поверхностном слое раствора оказывается больше, чем в глубине раствора.
Количественной мерой адсорбции на границе раствор-пар является поверхностный избыток Г (гамма), равный числу молей растворенного вещества в поверхностном слое. Количественное соотношение между адсорбцией (поверхностным избытком) растворенного вещества и изменением поверхностного натяжения раствора с ростом концентрации раствора определяет изотерма адсорбции Гиббса:
![]() (IV.5)
(IV.5)
График изотермы адсорбции ПАВ представлен на рис. 4.2. Из уравнения (IV.5) следует, что направление процесса – концентрирование вещества в поверхностном слое или, наоборот, нахождение его в объеме жидкой фазы – определяется знаком производной dσ/dС. Отрицательная величина данной производной соответствует накоплению вещества в поверхностном слое (Г > 0), положительная – меньшей концентрации вещества в поверхностном слое по сравнению с его концентрацией в объеме раствора.
Величину g = –dσ/dС называют также поверхностной активностью растворенного вещества. Поверхностную активность ПАВ при некоторой концентрации С1 определяют графически, проводя касательную к изотерме поверхностного натяжения в точке С = С1; при этом поверхностная активность численно равна тангенсу угла наклона касательной к оси концентраций:
![]() (IV.6)
(IV.6)
Нетрудно заметить, что с ростом концентрации поверхностная активность ПАВ уменьшается. Поэтому поверхностную активность вещества обычно определяют при бесконечно малой концентрации раствора; в этом случае её величина, обозначаемая gо, зависит только от природы ПАВ и растворителя. Исследуя поверхностное натяжение водных растворов органических веществ, Траубе и Дюкло установили для гомологических рядов поверхностно-активных веществ следующее эмпирическое правило:
В любом гомологическом ряду при малых концентрациях удлинение углеродной цепи на одну группу СН2 увеличивает поверхностную активность в 3 – 3.5 раза.
Для водных растворов жирных кислот зависимость поверхностного натяжения от концентрации описывается эмпирическим уравнением Шишковского:
![]() (IV.6a)
(IV.6a)
Здесь b и K – эмпирические постоянные, причём значение b одинаково для всего гомологического ряда, а величина К увеличивается для каждого последующего члена ряда в 3 – 3,5 раза.
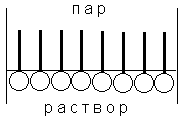
Рис. 4.3 Предельная ориентация
молекул ПАВ в поверхностном слое
Молекулы большинства ПАВ обладают дифильным строением, т.е. содержат как полярную группу, так и неполярный углеводородный радикал. Расположение таких молекул в поверхностном слое энергетически наиболее выгодно при условии ориентации молекул полярной группой к полярной фазе (полярной жидкости), а неполярной – к неполярной фазе (газу или неполярной жидкости). При малой концентрации раствора тепловое движение нарушает ориентацию молекул ПАВ; при повышении концентрации происходит насыщение адсорбционного слоя и на поверхности раздела фаз образуется слой "вертикально" ориентированных молекул ПАВ (рис. 4.3). Образование такого мономолекулярного слоя соответствует минимальной величине поверхностного натяжения раствора ПАВ и максимальному значению адсорбции Г (рис. 4.1-4.2); при дальнейшем увеличении концентрации ПАВ в растворе поверхностное натяжение и адсорбция не изменяются.
1.3 Адсорбция на границе твердое тело – газ
При адсорбции газов на твердых телах описание взаимодействия молекул адсорбата и адсорбента представляет собой весьма сложную задачу, поскольку характер их взаимодействия, определяющий характер адсорбции, может быть различным. Поэтому обычно задачу упрощают, рассматривая два крайних случая, когда адсорбция вызывается физическими или химическими силами соответственно физическую и химическую адсорбцию.
Физическая адсорбция возникает за счет ван-дер-ваальсовых взаимодействий. Она характеризуется обратимостью и уменьшением адсорбции при повышении температуры, т.е. экзотермичностью, причем тепловой эффект физической адсорбции обычно близок к теплоте сжижения адсорбата (10 – 80 кДж/моль). Таковой является, например, адсорбция инертных газов на угле.
Химическая адсорбция (хемосорбция) осуществляется путем химического взаимодействия молекул адсорбента и адсорбата. Хемосорбция обычно необратима; химическая адсорбция, в отличие от физической, является локализованной, т.е. молекулы адсорбата не могут перемещаться по поверхности адсорбента. Так как хемосорбция является химическим процессом, требующим энергии активации порядка 40 – 120 кДж/моль, повышение температуры способствует её протеканию. Примером химической адсорбции является адсорбция кислорода на вольфраме или серебре при высоких температурах.
Следует подчеркнуть, что явления физической и химической адсорбции чётко различаются в очень редких случаях. Обычно осуществляются промежуточные варианты, когда основная масса адсорбированного вещества связывается сравнительно слабо и лишь небольшая часть – прочно. Например, кислород на металлах или водород на никеле при низких температурах адсорбируются по законам физической адсорбции, но при повышении температуры начинает протекать химическая адсорбция. При повышении температуры увеличение химической адсорбции с некоторой температуры начинает перекрывать падение физической адсорбции, поэтому температурная зависимость адсорбции в этом случае имеет четко выраженный минимум (рис. 4.4).
Рис. 4.4 Зависимость объема
адсорбированного никелем водорода от температуры
При постоянной температуре количество адсорбированного вещества зависит только от равновесных давления либо концентрации адсорбата; уравнение, связывающее эти величины, называется изотермой адсорбции.
4.1.4 Теории адсорбции
Единой теории, которая достаточно корректно описывала бы все виды адсорбции на разных поверхностях раздела фаз, не имеется; рассмотрим поэтому некоторые наиболее распространенные теории адсорбции, описывающие отдельные виды адсорбции на поверхности раздела твердое тело – газ или твердое тело – раствор.
Теория мономолекулярной адсорбции Ленгмюра
Теория мономолекулярной адсорбции, которую разработал американский химик И. Ленгмюр, основывается на следующих положениях.
1) Адсорбция является локализованной и вызывается силами, близкими к химическим.
2) Адсорбция происходит не на всей поверхности адсорбента, а на активных центрах, которыми являются выступы либо впадины на поверхности адсорбента, характеризующиеся наличием т.н. свободных валентностей. Активные центры считаются независимыми (т.е. один активный центр не влияет на адсорбционную способность других), и тождественными.
3) Каждый активный центр способен взаимодействовать только с одной молекулой адсорбата; в результате на поверхности может образоваться только один слой адсорбированных молекул.
4) Процесс адсорбции является обратимым и равновесным – адсорбированная молекула удерживается активным центром некоторое время, после чего десорбируется; т.о., через некоторое время между процессами адсорбции и десорбции устанавливается динамическое равновесие.
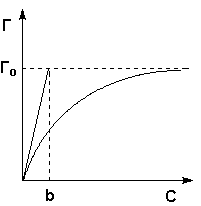
Рис. 4.5 Изотерма
мономолекулярной адсорбции
В состоянии равновесия скорость адсорбции равна скорости десорбции. Скорость десорбции прямо пропорциональна доле занятых активных центров (х), а скорость адсорбции прямо пропорциональна произведению концентрации адсорбата на долю свободных активных центров (1 – х):
![]() (IV.7)
(IV.7)
![]() (IV.8)
(IV.8)
![]() (IV.9)
(IV.9)
Отсюда находим х:
![]() (IV.10)
(IV.10)
Разделив числитель и знаменатель правой части уравнения (IV.10) на kA, получим:
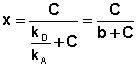 (IV.11)
(IV.11)
Максимально возможная величина адсорбции Го достигается при условии, что все активные центры заняты молекулами адсорбата, т.е. х = 1. Отсюда следует, что х = Г / Го. Подставив это в уравнение (IV.11), получаем:
![]() (IV.12)
(IV.12)
![]() (IV.13)
(IV.13)
Уравнение (IV.13) есть изотерма мономолекулярной адсорбции, связывающая величину адсорбции Г с концентрацией адсорбата С. Здесь b – некоторая постоянная для данной пары адсорбент-адсорбат величина (отношение констант скоростей десорбции и адсорбции), численно равная концентрации адсорбата, при которой занята половина активных центров. График изотермы адсорбции Ленгмюра приведен на рис. 4.5. Константу b можно определить графически, проведя касательную к изотерме адсорбции в точке С = 0.
При описании процесса адсорбции газов в уравнении (IV.13) концентрация может быть заменена пропорциональной величиной парциального давления газа:
![]() (IV.14)
(IV.14)
Теория мономолекулярной адсорбции Ленгмюра применима для описания некоторых процессов адсорбции газов и растворенных веществ при небольших давлениях (концентрациях) адсорбата.
Теория полимолекулярной адсорбции Поляни
На практике часто (особенно при адсорбции паров) встречаются т.н. S-образные изотермы адсорбции (рис. 4.6), форма которых свидетельствует о возможном, начиная с некоторой величины давления, взаимодействии адсорбированных молекул с адсорбатом.
Рис. 4.6 Изотерма полимолекулярной адсорбции
Для описания таких изотерм адсорбции М. Поляни предложил теорию полимолекулярной адсорбции, основанную на следующих основных положениях:
1. Адсорбция вызвана чисто физическими силами.
2. Поверхность адсорбента однородна, т.е. на ней нет активных центров; адсорбционные силы образуют непрерывное силовое поле вблизи поверхности адсорбента.
3. Адсорбционные силы действуют на расстоянии, большем размера молекулы адсорбата. Иначе говоря, у поверхности адсорбента существует некоторый адсорбционный объём, который при адсорбции заполняется молекулами адсорбата.
4. Притяжение молекулы адсорбата поверхностью адсорбента не зависит от наличия в адсорбционном объеме других молекул, вследствие чего возможна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры и, следовательно, с изменением температуры адсорбционный объем не меняется.
Уравнение Фрейндлиха
Теоретические представления, развитые Ленгмюром и Поляни, в значительной степени идеализируют и упрощают истинную картину адсорбции. На самом деле поверхность адсорбента неоднородна, между адсорбированными частицами имеет место взаимодействие, активные центры не являются полностью независимыми друг от друга и т.д. Все это усложняет вид уравнения изотермы. Г. Фрейндлих предположил, что число молей адсорбированного газа или растворенного вещества, приходящееся на единицу массы адсорбента (т.н. удельная адсорбция x/m) должна быть пропорциональна равновесному давлению (для газа) или равновесной концентрации (для веществ, адсорбируемых из раствора) адсорбента, возведенной в некоторую степень, которая всегда меньше единицы:
![]() (IV.15)
(IV.15)
![]() (IV.16)
(IV.16)
|
|
|
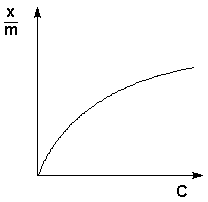
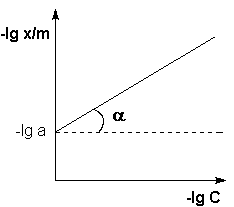
Рис. 4.7 Изотерма адсорбции Фрейндлиха в обычных (а) и логарифмических (б) координатах
Показатель степени n и коэффициент пропорциональности а в уравнении Фрейндлиха определяются экспериментально. Логарифмируя уравнения (IV.15 - IV.16), получаем:
![]() (IV.17)
(IV.17)
![]() (IV.18)
(IV.18)
Т.о., зависимость логарифма удельной адсорбции от логарифма концентрации (давления) графически выражается прямой линией, отсекающей на оси ординат отрезок, равный lga, тангенс угла наклона которой к оси абсцисс равен по величине показателю степени при давлении или концентрации (рис. 4.7):
![]() (IV.19)
(IV.19)
4.1.5 Адсорбция на границе твердое тело – раствор
Молекулярная адсорбция из растворов
Изотермы адсорбции растворенных веществ из раствора
по своему виду аналогичны изотермам адсорбции для газов; для разбавленных
растворов эти изотермы хорошо описываются уравнениями Фрейндлиха или Ленгмюра,
если в них подставить равновесную концентрацию растворенного вещества в
растворе. Однако адсорбция из растворов является значительно более сложным
явлением по сравнению с газовой, поскольку одновременно с адсорбцией
растворенного вещества часто происходит и адсорбция растворителя.
Рис. 4.8 Ориентация молекул ПАВ
на поверхности адсорбента
Зависимость адсорбции от строения молекул адсорбата очень сложна, и вывести какие-либо закономерности довольно трудно. Молекулы многих органических веществ состоят из полярной (гидрофильной) и неполярной (гидрофобной) группировок, т.е. являются поверхностно-активными веществами. Молекулы ПАВ при адсорбции на твердом адсорбенте ориентируются на его поверхности таким образом, чтобы полярная часть молекулы была обращена к полярной фазе, а неполярная – к неполярной. Так, при адсорбции алифатических карбоновых кислот из водных растворов на неполярном адсорбенте – активированном угле – молекулы ориентируются углеводородными радикалами к адсорбенту; при адсорбции из бензола (неполярный растворитель) на полярном адсорбенте силикагеле – ориентация молекул кислоты будет обратной (рис. 4.8).
Адсорбция из растворов электролитов
Адсорбция из водных растворов электролитов происходит, как правило, таким образом, что на твердом адсорбента из раствора адсорбируются преимущественно ионы одного вида. Преимущественная адсорбция из раствора или аниона, или катиона определяется природой адсорбента и ионов. Механизм адсорбции ионов из растворов электролитов может быть различным; выделяют обменную и специфическую адсорбцию ионов.
Обменная адсорбция представляет собой процесс обмена ионов между раствором и твердой фазой, при котором твердая фаза поглощает из раствора ионы какого-либо знака (катионы либо анионы) и вместо них выделяет в раствор эквивалентное число других ионов того же знака. Обменная адсорбция всегда специфична, т.е. для данного адсорбента к обмену способны только определенные ионы; обменная адсорбция обычно необратима.
При специфической адсорбции адсорбция на поверхности твердой фазы ионов какого-либо вида не сопровождается выделением в раствор эквивалентного числа других ионов того же знака; твердая фаза при этом приобретает электрический заряд. Это приводит к тому, что вблизи поверхности под действием сил электростатического притяжения группируется эквивалентное число ионов с противоположным зарядом, т.е. образуется двойной электрический слой. Взаимодействие концентрирующихся на поверхности зарядов приводит к понижению поверхностной энергии системы. Для случая специфической адсорбции электролита Песковым и Фаянсом было сформулировано следующее эмпирическое правило (правило Пескова – Фаянса):
На поверхности кристаллического твердого тела из раствора электролита специфически адсорбируется ион, который способен достраивать его кристаллическую решетку или может образовывать с одним из ионов, входящим в состав кристалла, малорастворимое соединение.
4.2 КОЛЛОИДНЫЕ СИСТЕМЫ
Коллоидные системы относятся к дисперсным системам – системам, где одно вещество в виде частиц различной величины распределено в другом (см. разд. 4.1). Дисперсные системы чрезвычайно многообразны; практически всякая реальная система является дисперсной. Дисперсные системы классифицируют прежде всего по размеру частиц дисперсной фазы (или степени дисперсности); кроме того, их разделяют на группы, различающиеся по природе и агрегатному состоянию дисперсной фазы и дисперсионной среды.
Если дисперсионной средой является жидкость, а дисперсной фазой – твердые частицы, система называется взвесью или суспензией; если дисперсная фаза представляет собой капельки жидкости, то систему называют эмульсией. Эмульсии, в свою очередь, подразделяют на два типа: прямые, или "масло в воде" (когда дисперсная фаза – неполярная жидкость, а дисперсионная среда – полярная жидкость) и обратные, или "вода в масле" (когда полярная жидкость диспергирована в неполярной). Среди дисперсных систем выделяют также пены (газ диспергирован в жидкости) и пористые тела (твердая фаза, в которой диспергированы газ либо жидкость). Основные типы дисперсных систем приведены в табл.1.
По степени дисперсности выделяют обычно следующие классы дисперсных систем:
Грубодисперсные системы системы, размер частиц дисперсной фазы в которых превышает 10-7 м.
Коллоидные системы – системы, размер частиц дисперсной фазы в которых составляет 10-7 – 10-9 м. Коллоидные системы характеризуются гетерогенностью, т.е. наличием поверхностей раздела фаз и очень большим значением удельной поверхности дисперсной фазы. Это обусловливает значительный вклад поверхностной фазы в состояние системы и приводит к появлению у коллоидных систем особых, присущих только им, свойств.
Иногда выделяют молекулярно(ионно)-дисперсные системы, которые, строго говоря, являются истинными растворами, т.е. гомогенными системами, поскольку в них нет поверхностей раздела фаз.
Коллоидные системы, в свою очередь, подразделяются на две группы, резко отличные по характеру взаимодействий между частицами дисперсной фазы и дисперсионной среды – лиофобные коллоидные растворы (золи) и растворы высокомолекулярных соединений (ВМС), которые ранее называли лиофильными коллоидами. К лиофобным коллоидам относятся системы, в которых частицы дисперсной фазы слабо взаимодействуют с дисперсионной средой; эти системы могут быть получены только с затратой энергии и устойчивы лишь в присутствии стабилизаторов.
Растворы ВМС образуются самопроизвольно благодаря сильному взаимодействию частиц дисперсной фазы с дисперсионной средой и способны сохранять устойчивость без стабилизаторов. Лиофобные коллоиды и растворы ВМС различаются также и структурой частиц, составляющих дисперсную фазу. Для лиофобных коллоидов единицей структуры является сложный многокомпонентный агрегат переменного состава – мицелла, для растворов ВМС – макромолекула.
Таблица 1. Основные типы дисперсных систем
|
Дисперсная |
Дисперсионная среда |
Условное обозначение |
Примеры дисперсных систем |
| Жидкость | Газ | ж/г | Туман, облака, жидкие аэрозоли |
| Твердое тело | Газ | т/г | Дым, пыль, твердые аэрозоли |
| Газ | Жидкость | г/ж | Пены, газовые эмульсии |
| Жидкость | Жидкость | ж/ж | Эмульсии (молоко, латекс) |
| Твердое тело | Жидкость | т/ж | Суспензии, коллоидные растворы, гели, пасты |
| Газ | Твердое тело | г/т | Твердые пены, пористые тела (пенопласты, силикагель, пемза) |
| Жидкость | Твердое тело | ж/т | Жемчуг, опал |
| Твердое тело | Твердое тело | т/т | Цветные стекла, сплавы |
4.2.1 Методы получения лиофобных коллоидов
Коллоидные системы по степени дисперсности занимают промежуточное положение между истинными растворами (молекулярно- или ионно-дисперсными системами) и грубодисперсными системами. Поэтому коллоидные растворы могут быть получены либо путем ассоциации (конденсации) молекул и ионов истинных растворов, либо дальнейшим раздроблением частиц дисперсной фазы грубодисперсных систем.
Методы получения коллоидных растворов также можно разделить на две группы: методы конденсации и диспергирования (в отдельную группу выделяется метод пептизации, который будет рассмотрен позднее). Еще одним необходимым для получения золей условием, помимо доведения размеров частиц до коллоидных, является наличие в системе стабилизаторов – веществ, препятствующих процессу самопроизвольного укрупнения коллоидных частиц.
Дисперсионные методы
Дисперсионные методы основаны на раздроблении твердых тел до частиц коллоидного размера и образовании таким образом коллоидных растворов. Процесс диспергирования осуществляется различными методами: механическим размалыванием вещества в т.н. коллоидных мельницах, электродуговым распылением металлов, дроблением вещества при помощи ультразвука.
Методы конденсации
Вещество, находящееся в молекулярно-дисперсном состоянии, можно перевести в коллоидное состояние при замене одного растворителя другим – т.н. методом замены растворителя. В качестве примера можно привести получение золя канифоли, которая не растворяется в воде, но хорошо растворима в этаноле. При постепенном добавлении спиртового раствора канифоли к воде происходит резкое понижение растворимости канифоли, в результате чего образуется коллоидный раствор канифоли в воде. Аналогичным образом может быть получен гидрозоль серы.
Коллоидные растворы можно получать также и методом химической конденсации, основанном на проведении химических реакций, сопровождающихся образованием нерастворимых или малорастворимых веществ. Для этой цели используются различные типы реакций – разложения, гидролиза, окислительно-восстановительные и т.д. Так, красный золь золота получают восстановлением натриевой соли золотой кислоты формальдегидом:
NaAuO2 + HCOH + Na2CO3 ––> Au + HCOONa + H2O
Строение мицеллы данного золя можно представить следующей схемой (см. разд. 4.2.2):
{[Au]m· n AuO2–· (n-x) Na+}x– · xNa+
Аналогичным образом получают золь серебра из разбавленных растворов нитрата серебра. Золь серы может быть получен окислением сероводорода кислородом в водном растворе, действием на сероводород сернистого газа либо разложением тиосерной кислоты:
H2S + O2 ––> S + H2O
H2S + SO2 ––> S + H2O
H2S2O3 > H2O + SO2 + S
Строение золя серы можно представить схемой:
{[S]m · n HS– · (n-x) H+}x– · x H+
Золи могут быть получены также в результате реакций ионного обмена, в результате которых выделяется нерастворимая соль, образующая при определенных условиях коллоидный раствор; так получают, например, золь иодида серебра (см. ниже).
Процесс гидролиза различных солей может приводить к образованию коллоидных растворов нерастворимых гидроксидов или кислот; так получают, например, золь гидроксида железа(III), имеющий следующее строение:
{[Fe(OH)3]m n FeO+ · (n–x)Cl–}x+ x Cl–
4.2.2 Агрегативная устойчивость лиофобных коллоидов.
Строение коллоидной мицеллы
Лиофобные коллоиды обладают очень высокой поверхностной энергией и являются поэтому термодинамически неустойчивыми; это делает возможным самопроизвольный процесс уменьшения степени дисперсности дисперсной фазы (т.е. объединение частиц в более крупные агрегаты) – коагуляцию золей. Тем не менее золям присуща способность сохранять степень дисперсности – агрегативная устойчивость, которая обусловлена, во-первых, снижением поверхностной энергии системы благодаря наличию на поверхности частиц дисперсной фазы двойного электрического слоя и, во-вторых, наличием кинетических препятствий для коагуляции в виде электростатического отталкивания частиц дисперсной фазы, имеющих одноименный электрический заряд.
Строение структурной единицы лиофобных коллоидов – мицеллы может быть показано лишь схематически, поскольку мицелла не имеет определенного состава. Рассмотрим строение коллоидной мицеллы на примере гидрозоля иодида серебра, получаемого взаимодействием разбавленных растворов нитрата серебра и иодида калия:
AgNO3 + KI ––> AgI + KNO3
Коллоидная мицелла золя иодида серебра (см. рис. 4.9) образована микрокристаллом иодида серебра, который способен к избирательной адсорбции из окружающей среды катионов Ag+ или иодид-ионов. Если реакция проводится в избытке иодида калия, то кристалл будет адсорбировать иодид-ионы; при избытке нитрата серебра микрокристалл адсорбирует ионы Ag+. В результате этого микрокристалл приобретает отрицательный либо положительный заряд; ионы, сообщающие ему этот заряд, называются потенциалопределяющими, а сам заряженный кристалл – ядром мицеллы. Заряженное ядро притягивает из раствора ионы с противоположным зарядом – противоионы; на поверхности раздела фаз образуется двойной электрический слой. Некоторая часть противоионов адсорбируется на поверхности ядра, образуя т.н. адсорбционный слой противоионов; ядро вместе с адсорбированными на нем противоионами называют коллоидной частицей или гранулой. Остальные противоионы, число которых определяется, исходя из правила электронейтральности мицеллы, составляют диффузный слой противоионов; противоионы адсорбционного и диффузного слоев находятся в состоянии динамического равновесия адсорбции – десорбции.
Схематически мицелла золя иодида серебра, полученного в избытке иодида калия (потенциалопределяющие ионы – анионы I–, противоионы – ионы К+) может быть изображена следующим образом:
{[AgI]m · nI– · (n-x)K+}x– · x K+
При получении золя иодида серебра в избытке нитрата серебра коллоидные частицы будут иметь положительный заряд:
{[AgI]m · nAg+ · (n-x)NO3–}x+ · x NO3–

Рис. 4.9. Строение коллоидной мицеллы
Агрегативная устойчивость золей обусловлена, таким образом, рядом факторов: во-первых, снижением поверхностной энергии дисперсной фазы (т.е. уменьшения движущей силы коагуляции) в результате образования двойного электрического слоя и, во-вторых, наличием кинетических препятствий для коагуляции в виде электростатического отталкивания имеющих одноименный заряд коллоидных частиц и противоионов. Еще одна причина устойчивости коллоидов связана с процессом гидратации (сольватации) ионов. Противоионы диффузного слоя сольватированы; эта оболочка из сольватированных противоионов также препятствует слипанию частиц.
4.2.3 Коагуляция лиофобных коллоидов
Как было показано выше, лиофобные коллоиды являются термодинамически неустойчивыми системами, существующими благодаря стабилизации за счет возникновения двойного электрического слоя. Изменение состояния ДЭС может, следовательно, привести к потере агрегативной устойчивости – слипанию частиц в более крупные агрегаты, т.е. коагуляции золя. Коагуляция золей может быть вызвана различными факторами: прибавлением электролитов, нагреванием или замораживанием, механическим воздействием и т.д. Наиболее важным и изученным фактором коагуляции гидрофобных коллоидов является воздействие на них растворов электролитов.
Для коагуляции золей электролитами установлен ряд эмпирических закономерностей.
1. Для начала коагуляции золя необходима некоторая минимальная концентрация электролита, называемая порогом коагуляции γ.
2. Коагулирующим действием обладает тот из ионов электролита, заряд которого противоположен заряду коллоидных частиц, причем коагулирующее действие иона тем сильнее, чем больше его заряд (правило Шульце – Гарди или правило значности). Величины порогов коагуляции двухзарядных ионов примерно на порядок, а трехзарядных – на два порядка меньше, чем для однозарядных ионов. Правило значности имеет приближенный характер и справедливо только для неорганических ионов; некоторые однозарядные органические ионы обладают более сильным коагулирующим действием, чем двухзарядные неорганические ионы, что обусловлено их сильной специфической адсорбируемостью.
3. В рядах неорганических ионов с одинаковыми зарядами коагулирующее действие возрастает с уменьшением гидратируемости ионов; например, в ряду однозарядных катионов щелочных металлов коагулирующее действие возрастает от лития к рубидию:
γ (Li+) > γ (Na+) > γ (К+) > γ (Rb+)
Ряды, в которые сгруппированы по возрастанию либо по убыванию коагулирующего действия ионы с одинаковым зарядом, называют лиотропными рядами.
4. В осадках, получаемых при коагуляции золей электролитами, всегда присутствуют ионы, вызвавшие коагуляцию.
5. При коагуляции золей смесями электролитов сравнительно редко наблюдается их независимое (аддитивное) действие; обычно имеет место взаимное усиление либо ослабление коагулирующего действия (синергизм либо антагонизм ионов).
Механизм и кинетика коагуляции золей электролитами
Необходимому для коагуляции сближению частиц дисперсной фазы препятствует, как было показано выше, электростатическое отталкивание имеющих одноименный заряд коллоидных частиц и противоионов и взаимодействие сольватных оболочек противоионов диффузного слоя. При добавлении к золю раствора электролита имеющееся равновесие адсорбции – десорбции между противоионами адсорбционного и диффузного слоев смещается в сторону адсорбции вследствие увеличения в дисперсионной среде концентрации ионов, имеющих заряд, противоположный заряду ядра (ионы с одноименным зарядом в равновесии адсорбции – десорбции не участвуют). Адсорбция дополнительного числа противоионов приводит к уменьшению заряда коллоидных частиц, уменьшению числа противоионов диффузного слоя (уменьшению толщины ДЭС) и, следовательно, к снижению агрегативной устойчивости золя. При достижении некоторого предельного значения заряда коллоидные частицы получают возможность сближения и объединения в более крупные агрегаты за счет ван-дер-ваальсовых сил; иными словами, происходит коагуляция золя.
Очевидно, что, поскольку при адсорбции многозарядных противоионов заряд коллоидной частицы уменьшается быстрее, чем при адсорбции того же числа однозарядных противоионов; адсорбируемость неорганических ионов с увеличением их заряда также возрастает. Следствием этого и является тот факт, что величина порога коагуляции для неорганических ионов будет тем меньше, чем больше заряд иона-коагулянта (величина порога коагуляции γ обратно пропорциональна заряду иона-коагулянта в шестой степени z6).
Процесс коагуляции золя характеризуется определенной
величиной скорости коагуляции, которую можно определить как изменение числа
коллоидных частиц в единице объема за единицу времени. Скорость коагуляции золя
электролитами зависит как от концентрации самого золя, так и от концентрации
электролитов. Типичный вид коагуляционной кривой (зависимости отношения
концентрации коллоидных частиц n к их начальной концентрации nо от
времени t) и кривой зависимости скорости коагуляции V от концентрации
электролита С показан на рисунках 4.10-4.11. На кривой ОАБВ (рис. 4.11) отрезок
ОА отвечает периоду скрытой коагуляции, при которой золь сохраняет свою
устойчивость. В точке А при концентрации электролита С1 начинается явная
коагуляция; на участке АБ скорость коагуляции быстро возрастает с ростом
концентрации электролита. На участке БВ скорость коагуляции остается
постоянной; это связано с тем, что при концентрации электролита С2
величина ζ-потенциала становится равной нулю; скорость коагуляции при этом
достигает максимального значения.
Рис. 4.10 Коагуляционная кривая. Рис. 4.11 Зависимость скорости коагуляции от концентрации.
Взаимная коагуляция золей
Коагуляция золя может быть вызвана его взаимодействием с другим золем, частицы которого имеют противоположный заряд. Так, смешение золя гидроксида железа, частицы которого имеют положительный заряд, с отрицательно заряженным золем сульфида мышьяка приводит к их взаимной коагуляции:
{[Fe(OH)3]m n FeO+· (n-x)Cl–}x+ xCl–{[Аs2S3]m · n НS–· (n-x)Н+}x– · xН+
В данном случае коагуляция обусловлена тем, что коллоидные частицы одного вида являются как бы очень крупными многозарядными ионами – коагулянтами для частиц другого вида. Взаимная коагуляция коллоидных систем может наблюдаться и тогда, когда частицы золей имеют одноименный заряд; в этом случае причиной потери устойчивости одного из золей является сильная специфическая адсорбция иона – стабилизатора данной системы поверхностью коллоидных частиц другой системы.
Старение золей и пептизация
Термодинамическая неустойчивость лиофобных коллоидных систем является причиной старения золей – самопроизвольной коагуляции (автокоагуляции) золей. Автокоагуляция золей происходит значительно медленнее, чем коагуляция электролитами; так, золи золота могут сохраняться без видимых изменений десятилетиями. Одной из основных причин старения золей является медленно совершающийся процесс перекристаллизации вещества ядра.
Пептизацией (дезагрегацией) называется процесс расщепления коагулировавшего золя (коагулята) на первичные частицы – процесс, противоположный коагуляции. Пептизация возможна лишь тогда, когда структура частиц в коагуляте не изменена по сравнению с первоначальной (т.е. когда еще не произошло полного сращивания частиц и они слабо связаны друг с другом). Различают непосредственную и опосредованную пептизацию.
Непосредственная пептизация происходит в результате добавления к коагуляту электролита, содержащего потенциалопределяющий ион; в результате его специфической адсорбции на поверхности частиц дисперсной фазы их заряд вновь увеличивается, толщина двойного электрического слоя возрастает. Это приводит к тому, что силы отталкивания между частицами начинают преобладать над силами притяжения; происходит деагрегация – распад образовавшегося ранее агрегата из слипшихся частиц.
Опосредованная пептизация вызывается добавлением в систему вещества, химическое взаимодействие которого с поверхностью коагулята приводит к высвобождению потенциалопределяющих ионов. Например, коагулировавший золь гидроксида железа(III) может быть пептизирован добавлением в систему либо какой-либо соли железа (непосредственная пептизация), либо соляной кислоты (опосредованная пептизация).
4.2.4 Двойной электрический слой и электрокинетические явления
При рассмотрении строения мицеллы было показано, что на поверхности лиофобных коллоидов образуется двойной электрический слой. Первая теория строения ДЭС была развита Гельмгольцем и Перреном; в их представлении двойной электрический слой подобен плоскому конденсатору, внутренняя обкладка которого находится в твердой фазе, а внешняя – в жидкости параллельно поверхности ядра на расстоянии порядка диаметра иона. Потенциал электрического поля внутри ДЭС φ в этом случае линейно уменьшается с увеличением расстояния от поверхности r (рис. 4.12а).
Позднее Гуи и Чепмен предложили другую модель, согласно которой противоионы, благодаря тепловому движению, образуют вблизи твердой поверхности ядра диффузную ионную атмосферу. Уменьшение электрического потенциала ДЭС φ с увеличением расстояния r в этом случае происходит нелинейно (рис. 4.12б).
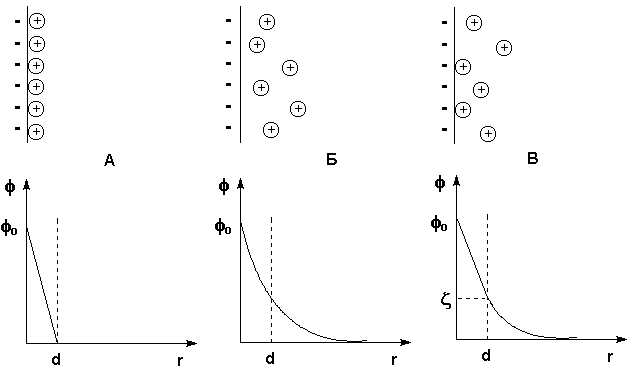
Рис. 4.12 Строение ДЭС: а) – по Гельмгольцу и Перрену, б) – по Гуи и Чепмену, в) – по Штерну. Вверху – схема расположения противоионов, внизу – зависимость потенциала от расстояния
Предложенная Штерном модель строения ДЭС объединяет ранние модели, учитывая как адсорбцию противоионов, так и их тепловое движение. Согласно этой модели, являющейся в настоящее время общепринятой, часть противоионов находится на расстояниях порядка диаметра иона от поверхности ядра, образуя т.н. слой Гельмгольца (адсорбционный слой противоионов), а другая часть образует диффузный слой (т.н. слой Гуи). Потенциал диффузной части двойного электрического слоя называют электрокинетическим потенциалом (см. рис.4.12в). Электрокинетический потенциал обычно обозначают греческой буквой ζ (дзета) и называют поэтому дзета-потенциалом. Поскольку ζ-потенциал пропорционален заряду коллоидной частицы, агрегативная устойчивость золя пропорциональна его величине.
Если поместить золь в постоянное электрическое поле, то, как и в растворах электролитов, заряженные частицы будут двигаться к противоположно заряженным электродам: коллоидная частица с адсорбированными на ней противоионами – в одну сторону, противоионы диффузного слоя – в другую. Сила, с которой электрическое поле действует на частицы и, следовательно, скорость движения частиц, очевидно, будет пропорциональна ζ-потенциалу. Движение частиц дисперсной фазы в электрическом поле называется электрофорезом. Явление электрофореза можно наблюдать, поместив в U-образную трубку какой-либо окрашенный золь, поверх которого налит не смешивающийся с золем бесцветный электролит. Если опустить в электролит электроды и наложить разность потенциалов, то граница окрашенного золя в одном из колен трубки будет подниматься, в другом – опускаться (рис. 4.13). Если поместить в U-образную трубку пористую перегородку (например, мелкий кварцевый песок) и заполнить е водой, то при наложении разности потенциалов в одном колене будет наблюдаться подъем уровня жидкости, в другом – его опускание (рис. 4.14). Движение дисперсной среды в электрическом поле относительно неподвижной дисперсной фазы (в рассмотренном случае – относительно поверхности пористых тел) называется электроосмосом. Явления электрофореза и электроосмоса получили общее название электрокинетических явлений.
|
|
|
|
Рис. 4.13
Схема опыта по электрофорезу |
Рис. 4.14 Схема
опыта по электроосмосу |
Скорость движения частиц дисперсной фазы при электрофорезе, а также скорость движения дисперсной среды при электроосмосе прямо пропорциональны напряженности электрического поля E и диэлектрической проницаемости дисперсионной среды ε и обратно пропорциональны вязкости среды η. Скорость движения частиц дисперсной фазы при электрофорезе U связана с величиной ζ-потенциала уравнением Гельмгольца-Смолуховского (К – постоянная, зависящая от формы частиц дисперсной фазы; для сферических частиц К = 6):
![]() (IV.20)
(IV.20)
Обратные электрофорезу и электроосмосу электрокинетические явления (т.н. электрокинетические явления второго рода) называются соответственно потенциал седиментации и потенциал протекания. Потенциал седиментации (эффект Дорна) – возникновение разности потенциалов при вынужденном движении дисперсной фазы относительно неподвижной дисперсионной среды (например, под действием силы тяжести). Потенциал протекания (эффект Квинке) есть явление возникновения разности потенциалов при движении дисперсионной среды относительно неподвижной дисперсной фазы (например, при продавливании электролита через пористое тело).
4.2.5 Кинетическая устойчивость золей. Седиментация
Частицы дисперсной фазы одновременно испытывают действие силы земного притяжения и архимедовой силы; в зависимости от соотношения плотностей дисперсионной среды и дисперсной фазы равнодействующая этих сил будет вынуждать частицы к оседанию либо всплытию. Процесс оседания либо всплытия коллоидных частиц в золе называется седиментацией. Однако седиментации всегда противодействует другой процесс, стремящийся к равномерному распределению коллоидных частиц по всему объему раствора – диффузия, осуществляемая под действием броуновского движения частиц. Соотношение между этими двумя процессами определяет кинетическую устойчивость золей способность коллоидных частиц удерживаться во взвешенном состоянии, не подвергаясь седиментации.
В статистической теории броуновского движения, развитой А. Эйнштейном, вводится понятие средний сдвиг ±Δx, представляющий собой проекцию расстояния между положениями частицы X1 и X2, в которых частица находилась во время двух последовательных наблюдений через время t. Значение квадрата среднего сдвига можно найти по уравнению Эйнштейна, связывающего Δx2 с температурой T, радиусом взвешенных частиц r и вязкостью среды η:
![]() (IV.21)
(IV.21)
Средний сдвиг частицы связан с коэффициентом диффузии D, который может быть рассчитан по уравнению (IV.22):
![]() (IV.22)
(IV.22)
![]() (IV.23)
(IV.23)
Как видно из уравнения (IV.23), величина
коэффициента диффузии определяется отношением тепловой энергии молекул kT и вязкостного
сопротивления диффузии со стороны среды. Поскольку процесс диффузии проявляется
тем сильнее, чем меньше масса коллоидных частиц, более крупные частицы оседают
либо всплывают в первую очередь. Кинетическая устойчивость золя, таким образом,
прямо пропорциональна степени дисперсности золя. Заметное оседание частиц в
системе, обладающей высокой кинетической устойчивостью, можно вызвать при
помощи центрифугирования золя, используя значительные по величине центробежные
силы, что многократно увеличивает силу, действующую на частицу и способствующую
её оседанию (современные ультрацентрифуги работают при ускорениях свыше
400000g).
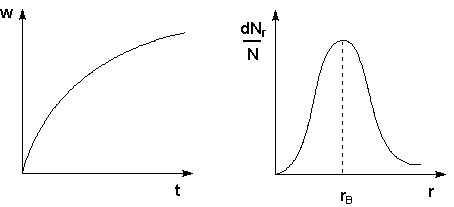
Рис. 4.15 Кривая седиментации Рис. 4.16 Кривая распределения
Методы седиментации и ультрацентрифугирования применяются для изучения полидисперсности коллоидных систем, обусловленной существованием в коллоидных системах частиц различных размеров. Изучение полидисперсности коллоидных систем для установления количественного распределения частиц по размерам (т.н. кривых распределения) – седиментационный анализ – производится при помощи измерения возрастания веса осевших частиц w со временем. По результатам такого исследования строят кривые седиментации (рис. 4.15). Проводя анализ кривой седиментации, можно рассчитать кривую распределения для данной системы, которая характеризует относительное содержание в системе частиц разного размера (рис. 4.16). Обычно кривые распределения содержат один максимум, который соответствует rв – наиболее вероятному радиусу частиц дисперсной фазы
4.2.6 Очистка коллоидных систем
Некоторые молекулярно-кинетические свойства
коллоидных систем используют для очистки золей от электролитов и молекулярных
примесей, которыми полученные золи часто бывают загрязнены. Наиболее
распространенными методами очистки коллоидных систем являются диализ, электродиализ
и ультрафильтрация, основанные на свойстве некоторых материалов – т.н.
полупроницаемых мембран (коллодия, пергамента, целлофана и т.п.) – пропускать
ионы и молекулы небольших размеров и задерживать коллоидные частицы. Все
полупроницаемые мембраны представляют собой пористые тела, и непроницаемость их
для коллоидных частиц обусловлена тем, что коэффициент диффузии для коллоидных
частиц значительно (на несколько порядков) меньше, чем для ионов и молекул,
имеющих намного меньшие массу и размеры.Прибор для очистки золей методом диализа
называется диализатором; простейший диализатор представляет собой сосуд, нижнее
отверстие которого затянуто полупроницаемой мембраной (рис. 4.17). Золь
наливают в сосуд и помещают последний в ёмкость с дистиллированной водой
(обычно проточной); ионы и молекулы примесей диффундируют через мембрану в
растворитель.
|
|
|
|
Рис. 4.17 Схема диализатора |
Рис. 4.18 Схема электродиализатора |
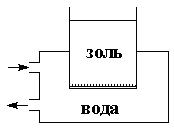
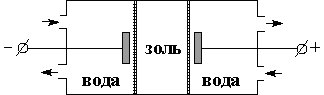
Диализ является очень медленным процессом; для более быстрой и полной очистки золей применяют электродиализ. Электродиализатор состоит из трех частей; в среднюю часть, отделенную от двух других полупроницаемыми мембранами, за которыми помещены электроды, наливается золь (рис. 4.18). При подключении к электродам разности потенциалов катионы содержащихся в золе электролитов диффундируют через мембрану к катоду, анионы к аноду. Преимущество электродиализа заключается в возможности удаления даже следов электролитов (необходимо помнить, что степень очистки ограничивается устойчивостью коллоидных частиц; удаление из золя ионов-стабилизаторов приведет к коагуляции).
Еще одним методом очистки золей является ультрафильтрация – отделение дисперсной фазы от дисперсионной среды путем фильтрования под давлением через полупроницаемые мембраны. При ультрафильтрации коллоидные частицы остаются на фильтре (мембране).
4.2.7 Оптические свойства коллоидных систем
Особые оптические свойства коллоидных растворов обусловлены их главными особенностями: дисперсностью и гетерогенностью. На оптические свойства дисперсных систем в значительной степени влияют размер и форма частиц. Прохождение света через коллоидный раствор сопровождается такими явлениями, как поглощение, отражение, преломление и рассеяние света. Преобладание какого-либо из этих явлений определяется соотношением между размером частиц дисперсной фазы и длиной волны падающего света. В грубодисперсных системах в основном наблюдается отражение света от поверхности частиц. В коллоидных растворах размеры частиц сравнимы с длиной волны видимого света, что предопределяет рассеяние света за счёт дифракции световых волн.
Светорассеяние в коллоидных растворах проявляется в виде опалесценции – матового свечения (обычно голубоватых оттенков), которое хорошо заметно на тёмном фоне при боковом освещении золя. Причиной опалесценции является рассеяние света на коллоидных частицах за счёт дифракции. С опалесценцией связано характерное для коллоидных систем явление – эффект Тиндаля: при пропускании пучка света через коллоидный раствор с направлений, перпендикулярных лучу, наблюдается образование в растворе светящегося конуса.
Процесс дифракционного светорассеяния на частицах, размер которых значительно меньше длины волны описывается уравнением Рэлея, связывающим интенсивность рассеянного единицей объёма света I с числом частиц в единице объёма ν, объёмом частицы V, длиной волны λ и амплитудой А падающего излучения и показателями преломления дисперсной фазы и дисперсионной среды n1 и n2 соответственно:
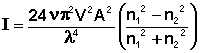 (IV.24)
(IV.24)
Из уравнения (IV.18) видно, что, чем меньше длина волны падающего излучения, тем больше будет рассеяние. Следовательно, если на частицу падает белый свет, наибольшее рассеивание рассеяние будут испытывать синие и фиолетовые компоненты. Поэтому в проходящем свете коллоидный раствор будет окрашен в красноватый цвет, а в боковом, отраженном – в голубой.
На сравнении интенсивности светорассеяния золей, один из которых имеет известную концентрацию (степень дисперсности), основан метод определения концентрации либо степени дисперсности золя, называемый нефелометрией. На использовании эффекта Тиндаля основывается ультрамикроскоп прибор, позволяющий наблюдать коллоидные частицы размером более 3 нанометров в рассеянном свете (в обычном микроскопе можно наблюдать частицы с радиусом не менее 200 нм из-за ограничений, связанных с разрешающей способностью оптики).
© 2009 База Рефератов