
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Учебное пособие: Принципы биохимических исследований
Учебное пособие: Принципы биохимических исследований
Содержание
Лекция 1. Оборудование биохимической лаборатории. Общие принципы биохимического исследования
Лекция 2. Разрушение клеток и экстракция. Центрифугирование
Лекция 3. Разделение белков путем осаждения
Лекция 4. Буферные растворы и специальные добавки. Ультрафильтрация. Диализ. Детергенты и их применение
Лекция 5. Общие принципы хроматографии, классификация хроматографических методов
Лекция 6. Материалы матриц сорбентов и обменников. Техника колоночной хроматографии
Лекция 7. Адсорбционная и распределительная хроматографии
Лекция 8. Тонкослойная хроматография
Лекция 9. Ионообменная хроматография
Лекция 10. Ионообменная ЖХВД белков. Хроматофокусирование
Лекция 11. Аффинная хроматография
Лекция 12. Гель-фильтрация
Лекция 13. Теоретические и методические основы электрофореза
Лекция 14. Изоэлектрическое фокусирование и изотахофорез
Лекция 15. Обнаружение, количественное определение и характеристика макромолекул после электрофореза
Лекция 16. Принцип иммунного электрофореза. Иммунофиксация
Лекция 17. Электросинерез. Электроиммуноанализ
Лекция 18. Методы меченых атомов
Лекция 19. Спектрофотометрические методы анализа
Лекция 20. Флюориметрические методы анализа
Лекция 21. Иммуноферментный анализ
Лекция 22. Радиометрический анализ. Масс-спектроскопия
Лекция 23. Блоттинг-анализ
Лекция 1. Оборудование биохимической лаборатории. Общие принципы биохимического исследования
Источники опасности и меры безопасности в лаборатории при проведении биохимического анализа. Особенности применения общих лабораторных методов в биохимическом эксперименте. Микро - и нанометоды.
Лабораторная посуда: материалы для её изготовления, выбор оптимального материала в зависимости от поставленной задачи биохимического эксперимента, виды лабораторной посуды, биосовместимые способы мытья и сушки лабораторной посуды, особые способы подготовки лабораторной посуды для биохимического анализа.
Исходные реактивы для биохимической лаборатории. Сведения о реактивах: маркировка реактивов, использование литературных и электронных источников справочной информации. Особенности хранения реактивов для биохимического анализа. Способы проверки качества и чистоты реактивов, выбор способа проверки, адекватного поставленной аналитической задаче. Методы дополнительной подготовки и очистки реактивов для биохимического анализа. Перекристаллизация.
Методы отбора реактивов в биохимическом анализе. Взвешивание: виды весов для аналитической биохимии, принципы и источники погрешностей взвешивания. Дозирование жидкостей, использование пипеточных дозаторов, возможные источники погрешностей. Особенности приготовления растворов в аналитической биохимии: принципы приготовления, способы выражения, концентраций, растворимости, растворители для биохимического анализа, способы постепенного добавления реактивов, растворение плохо растворимых веществ (суспендирование, эмульгирование, детергенты, использование которых допустимо в биохимическом анализе). Буферные растворы для использования в биохимическом анализе.
Методы контроля температуры в биохимической лабораторной практике.
Необходимость проведения ряда биохимических анализов в специальных условиях. Техника работ с реагентами, чувствительными к влаге, кислороды воздуха и свету. Проведение реакций в апротонных растворителях, в безводных условиях и в инертной атмосфере. Техника проведения фотохимических реакций.
Лекция 2. Разрушение клеток и экстракция. Центрифугирование
Принцип метода, основные определения и формулы. Центрифугирование применяется для разделения неоднородных жидких сред.
Центрифугирование позволяет разделить смесь, состоящую из двух или более компонентов с разной удельной плотностью, если по крайней мере один из этих компонентов - жидкость.
Разделение веществ с помощью центрифугирования основано на разном поведении частиц в центробежном поле. В центробежном поле частицы, имеющие разную плотность, форму или размеры, осаждаются с разной скоростью.
Скорость осаждения, или седиментации, зависит от центробежного ускорения (G), прямо пропорционального угловой скорости ротора (, в рад/с) и расстоянию между частицей и осью вращения (г, в см): G = 2 • г. Поскольку один оборот ротора составляет 2л радиан, угловую скорость ротора в оборотах в минуту (об. /мин) можно записать так: v = 2p60 (об. /мин), а центробежное ускорение тогда будет равно: G =4p2r/3600 (об. /мин) 2.
Центробежное ускорение обычно выражается в единицах g {гравитационная постоянная, равная 980 см*с-1) и называется относительным центробежным, ускорением (ОЦУ), т.е. ОЦУ=4p2r/3600*980 (об. /мин) 2 или ОЦУ = 1,11*10-5*r (об. /мин) 2 (*)
На основании уравнения (*) Доулом и Котциасом была составлена номограмма, выражающая зависимость ОЦУ от скорости вращения ротора и радиуса г - среднего радиуса вращения столбика жидкости в центрифужной пробирке (т.е. расстояния от оси вращения до середины столбика жидкости).
Номограмма для расчета центробежного ускорения

Для определения G соединяют прямой линией значения радиуса и скорости вращения ротора на крайних шкалах; точка пересечения этой прямой со средней шкалой дает искомую величину центробежного ускорения. Следует иметь в виду, что правая колонка цифр шкалы G соответствует правой колонке цифр шкалы скорости вращения ротора; левая - левой.
Скорость седиментации сферических частиц зависит не только от центробежного ускорения, но и от плотности и радиуса самих частиц и от вязкости среды суспендирования. Время осаждения сферической частицы в жидкой среде от мениска жидкости до дна центрифужной пробирки обратно пропорционально скорости седиментации и определяется следующим уравнением (закон Стокса, видоизмененный Сведбергом и Никольсом):
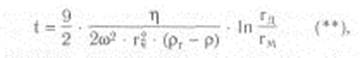
где t - время седиментации, с; h - вязкость среды, Паскаль • секунда; гч - радиус частицы, см; рч - плотность частицы (удельный вес); p - плотность среды (жидкости) или удельный вес; гм - расстояние от оси вращения до мениска жидкости, см; гд - расстояние от оси вращения до дна пробирки, см.
Как следует из уравнения (**), при заданной скорости вращения ротора время, необходимое для осаждения гомогенных сферических частиц, обратно пропорционально квадрату их радиусов и разности плотностей частиц и среды и прямо пропорционально вязкости среды. Поэтому смесь гетерогенных, приблизительно сферических частиц, различающихся по плотности и (или) размерам, можно выделить либо за счет разного времени осаждения их на дно пробирки при данном ускорении, либо за счет распределения седиментирующих частиц вдоль пробирки, устанавливающегося через определенный промежуток времени. При разделении веществ необходимо учитывать и такие важные факторы, как плотность и вязкость среды.
Описанными методами можно выделять клеточные органеллы из гомогенатов тканей. Основные компоненты клетки осаждаются в следующей последовательности: сначала целые клетки и их фрагменты, затем ядра, хлоропласты, митохондрии, лизосомы (или другие микротельца), микросомы (фрагменты гладкой и шероховатой эндоплазматической сети) и, наконец, рибосомы.
Осаждение несферических частиц не подчиняется уравнению (**), поэтому частицы одинаковой массы, но различной формы осаждаются при разных скоростях. Эта особенность используется при исследовании конформации макромолекул.
Препаративное центрифугирование заключается в выделении биологического материала для последующих биохимических исследований.
С помощью препаративного центрифугирования выделяют большое количество клеточных частиц для изучения их морфологии, структуры и биологической активности. Метод применяется для выделения таких биологических макромолекул, как ДНК и белки, из предварительно очищенных препаратов.
Аналитическое центрифугирование применяется главным образом для изучения чистых и практически чистых препаратов макромолекул или частиц, например, рибосом. В данном случае используется небольшое количество материала, а седиментация исследуемых частиц непрерывно регистрируется с помощью специальных оптических систем. Метод позволяет получать данные о чистоте, молекулярной массе и структуре материала.
В практике препаративное центрифугирование применяется гораздо чаще, чем аналитическое, поэтому мы остановимся на нем более подробно, хотя в основе обоих методов лежат общие принципы.
Лекция 3. Разделение белков путем осаждения
Осаждение нуклеиновых кислот.
Обычно, когда говорят о высаливании, имеют в виду высаливание именно сульфатом аммония. Этому методу уже более 130 лет. Раньше он применялся и для фракционирования, сейчас, в основном, как дешёвый и удобный метод осаждения белков. Можно считать, что повезло, если при этом получается ещё и существенная очистка (при высаливании из клеточного экстракта можно рассчитывать на приблизительно 2-10 кратное обогащение).
Почему именно сульфат аммония?
При равной молярной концентрации поливалентные анионы долее эффективны для высаливания, чем моновалентные (отчасти из-за того, что имеет значение не концентрация соли, а ионная сила раствора), а поливалентные катионы даже препятствуют действию поливалентных анионов. Получается, что оптимально сочетание - это поливалентный анион с моновалентными катионами.
Эффективность высаливания убывает в серии Гофмейстера (Hofmeister):
Цитрат > Сульфат > Фосфат > Хлорид > Нитрат > Тиоционат
В этом же ряду убывает стабилизирующий эффект и возрастают хаотропные свойства соли. Таким образом, наиболее подходящие кандидаты: цитрат и сульфат. Сульфат более удобен из-за лучшей растворимости (например, при нормальной температуре растворимость аммонийных солей цитрата и сульфата равны примерно 2.5М и 4.1М); низкой цены и стабилизирующего влияния, которое он оказывает на большинство белков при концентрациях выше 0.5М.
(NH4) 2SO4 преципитируют белки по двум механизмам
сульфат-ионы делают молекулу белка более компактной (менее растворимой) за счёт взаимодействия с положительно заряженными аминокислотами. Это взаимодействие более эффеективно при pH<pI.
обезвоживание. Один ион SO42 - имеет 13-14 молекул H2O только в первом гидратном слое и, возможно, больше - во втором. Если одна молекула сульфата координирует даже 15 молекул H2O, то 3M сульфат аммония связывает 45 из имеющихся в воде 55M молекул H2O.
I = 1/2 ![]() ci (zi) 2,
ci (zi) 2,
где:
ci - концентрация иона; zi - заряд иона.
Например, для 1M NaCl: I = 1/2 (1 (1) 2 + 1 (1) 2) = 1 для 1M (NH4) 2SO4: I = 1/2 (2 (1) 2 + 1 (1) 2) = 3
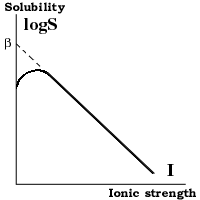
Растворимость белков
Влияние ионной силы и температуры При низкой ионной силе (<0.2M) растворимость белка увеличивается при повышении концентрации соли, так как экранирование притяжения противоположно заряженных групп приводит к разрыхлению структуры белка. При высокой ионной силе (>0.2M) - растворимость белка понижается из за высаливания и обезвоживания. Она падает экспоненциально с повышением ионной силы: logS = ß - KsI, где:
S [g/l] - растворимость белка; I - ионная сила; ß; Ks - константы.
Ks - слегка различается для различных белков и почти не зависит от pH и температуры. ß - сильно зависит от белка, pH и температуры; повышение температуры вызывает понижение ß => уменьшение растворимости белка.
Влияние pH. При низкой ионной силе (<0.2M) растворимость белка минимальна при pH равном изоэлектрической точке белка. При высоких концентрациях (NH4) 2SO4 растворимость повышается с повышением pH, так как при низких pH сульфат ион компактизует белок взаимодействуя с положительно заряженными группами. Так что лучше проводить осаждение при pH<pI.
Влияние начальной концентрации белка Белки бывают двух типов. Для типа I растворимость не зависит от исходной концентрации белка; для типа II - зависит сильно.
Высокая концентрация ионов аммония в осадке может мешать точному определению концентрации белка.
Высаливаются не только белки, но и, например, детергенты. Например, 0.5% Tween 20 и Triton X100 начинают агрегировать при концентрациях сульфата аммония больше 1M. Образующийся преципитат имеет плотность чуть меньше плотности солевого раствора. При центрифугировании он всплывает, прихватывая с собой белки.
Осаждение сульфатом амония нельзя использовать для белков, требующих присутствия Ca2+ из-за нерастворимости сульфата кальция.
Лекция 4. Буферные растворы и специальные добавки. Ультрафильтрация. Диализ. Детергенты и их применение
Буферные растворы (синоним: буферные смеси, буферные системы, буферы) - растворы с определенной концентрацией водородных ионов, содержащие сопряженную кислотно-основную пару, обеспечивающую устойчивость величины их водородного показателя при незначительных изменениях концентрации либо при добавлении небольшого количества кислоты или щелочи.
Кислотно-основная пара Б. р. представляет собой слабую кислоту и ее соль, образованную сильным основанием (например, уксусная кислота СН3СООН и ацетат натрия CH3COONa) или слабое основание и его соль, образованную сильной кислотой (например, гидроокись аммония NH4OH и хлористый аммоний NH4CI). При разведении раствора или добавлении к нему некоторого количества кислоты или щелочи кислотно-основная пара способна соответственно быть донором либо акцептором водородных ионов, поддерживая Т.о. величину водородного показателя на относительно постоянном уровне.
Буферные растворы сохраняют устойчивость буферных свойств в определенном интервале значений рН, то есть обладают определенной буферной емкостью. За единицу буферной емкости условно принимают емкость такого буферного раствора, для изменения рН которого на единицу требуется добавить 1 моль сильной кислоты или сильной щелочи на 1 л раствора. Буферная емкость находится в прямой зависимости от концентрации Б. р.: чем концентрированнее раствор, тем больше его буферная емкость; разведение Б. р. сильно уменьшает буферную емкость и лишь незначительно изменяет рН.
Тканевая жидкость, кровь, моча и другие биологические жидкости являются буферными растворами. Благодаря действию их буферных систем поддерживается относительное постоянство водородного показателя внутренней среды, обеспечивающее полноценность метаболических процессов. Наиболее важной буферной системой является бикарбонатная система крови. Концентрация в крови бикарбонатов служит одним из основных показателей кислотно-щелочного состояния организма. Этот показатель позволяет установить характер нарушения кислотно-щелочного равновесия при ряде патологических процессов.
В лабораторной практике Б. р. используют в тех случаях, когда то или иное исследование может быть проведено лишь при постоянном значении рН (например, определение активности ферментов, изучение кинетики ферментативных реакций, электрофоретическое разделение белковых смесей и др.) и в качестве стандартов при определении рН различных растворов, в т. ч. биологических жидкостей.
Буферные растворы готовят обычно путем растворения в воде взятых в соответствующих пропорциях слабой кислоты и ее соли, образованной щелочным металлом, частичной нейтрализации слабой кислоты сильной щелочью или слабого основания сильной кислотой, растворения смеси солей многоосновной кислоты.
Лекция 5. Общие принципы хроматографии, классификация хроматографических методов
Всем хроматографическим методам присущи некоторые общие характеристики, позволяющие ниже изложить элементы их обобщенной теории. Однако сначала рассмотрим специфические особенности различных вариантов хроматографического фракционирования. Это, с одной стороны, позволит за теоретическими рассуждениями все время видеть реальные черты хроматографического эксперимента, а с другой - даст возможность ввести классификацию хроматографиче-ских методов. В ходе дальнейшего изложения (в частности, для его разбиения по главам) удобнее всего классифицировать методы по основному принципу фракционирования. Такую классификацию мы рассмотрим достаточно подробно и лишь в конце раздела кратко отметим другие возможные варианты классификации.
КЛАССИФИКАЦИЯ ПО ПРИНЦИПУ ФРАКЦИОНИРОВАНИЯ.
Как уже упоминалось, в любом хроматографическом процессе фигурируют неподвижная и подвижная фазы, между которыми распределяются молекулы фракционируемой смеси веществ. Под основным принципом фракционирования будем подразумевать природу физического, химического или биологического явления, обусловливающего такое распределение.
КЛАССИФИКАЦИЯ ПО СПОСОБУ ЭЛЮЦИИ.
Фронтальный анализ.
Так называется вариант хроматографического процесса, когда раствор смеси компонентов непрерывно подается на вход хроматографической колонки. На выходе ее в этом случае появляются один за другим несколько "фронтов" элюата. За первым из них следует чистый, быстрее других мигрирующий в данной системе компонент смеси, отличающийся, очевидно, наименьшим сродством к неподвижной фазе. Второй фронт отмечает добавление к нему следующего по подвижности компонента. За третьим фронтом следует уже смесь трех компонентов. В настоящее время по вполне понятным причинам фронтальный анализ почти вышел из употребления и применяется лишь в отдельных, специальных случаях.
Вытеснительная хроматография.
В этом варианте в колонку или на стартовую линию хроматографической пластинки наносят определенную порцию раствора исходной смеси веществ, а затем ведут элюцию раствором вещества, обладающего заведомо большим сродством к неподвижной фазе хроматографической системы, чем любой из компонентов смеси. Происходит вытеснение их из неподвижной фазы, причем в первую очередь тех, которые обладают меньшим сродством к сорбенту, а затем и всех остальных. Элюент выталкивает все компоненты смеси впереди себя наподобие поршня. Так как они выходят в подвижную фазу концентрированными, то между ними также идет конкуренция за связь с неподвижной фазой. Компоненты, уступающие другим в силе сродства к этой фазе, оттесняются еще вперед, где сорбируются, но только до тех пор, пока их опять не вытеснят компоненты, обладающие большим сродством к сорбенту. В результате такого чередования сорбции и вытеснения компоненты смеси будут выходить из колонки один за другим в порядке возрастания силы их связи с неподвижной фазой. Ясно, что при этом зоны соседних компонентов будут соприкасаться или даже немного перекрываться друг с другом. Для аналитического фракционирования метод непригоден, но хорош для препаративного или полупромышленного разделения веществ, поскольку емкость колонки здесь используется очень эффективно.
Хроматографическая элюция.
В отличие от предыдущего в этом методе элюирующий раствор обладает меньшим сродством к сорбенту, чем любой из компонентов вносимой на колонку или пластинку смеси веществ. Эти компоненты постепенно "вымываются" из неподвижной фазы и движутся вдоль колонки за счет непрерывного перераспределения их молекул между неподвижной фазой и элюентом. Каждый из них мигрирует независимо от других в соответствии с соотношением сил его сродства к неподвижной и подвижной фазам. Миграция идет тем медленнее, чем больше сродство к неподвижной фазе. Именно этот, пригодный для аналитических целей вариант хроматографии подробно рассмотрен в следующем разделе, поэтому здесь можно ограничиться указанием на то, что при хроматографической элюции компоненты смеси выходят из колонки отдельными, разделенными друг от друга зонами, которые в соответствии с типичной формой профиля распределения вещества в каждой такой зоне (см. ниже) часто называют хроматографическими пиками.
КЛАССИФИКАЦИЯ ПО РАСПОЛОЖЕНИЮ НЕПОДВИЖНОЙ ФАЗЫ.
Эта классификация не требует особых пояснений. Если пористые гранулы геля или сорбента для любого типа хроматографии заполняют стеклянную или металлическую колонку, то говорят о хроматографии на колонке, или "колоночной хроматографии", хотя последнее выражение относится к категории укоренившегося жаргона. Хроматографию на колонке во многих случаях теперь ведут при очень большом давлении подачи элюента (до 300-400 атм), что позволяет уменьшить диаметр гранул до 5-10 мкм с вытекающими отсюда (см. ниже) существенными преимуществами в быстроте и качестве фракционирования микроколичеств исходного вещества. За это приходится расплачиваться использованием дорогостоящих стальных прецизионных колонок и специальной аппаратуры, но в случае серийных анализов такие затраты себя оправдывают. Жидкостную Хроматографию на колонках при высоком давлении условимся сокращенно обозначать ЖХВД. В английской литературе принято обозначение HPLC, которое расшифровывают как "high pressure (иногда - Align performance) liquid chromatography".
Если хроматографический процесс идет в наклонно расположенном, ровном и относительно толстом (несколько миллиметров), открытом с поверхности слое гранул, между которыми жидкость подвижной фазы течет только под действием силы тяжести, то его можно назвать хроматографией в толстом слое" Практически этот метод нашел себе применение только для гель-фильтрации.
Тонкий (0,1-0,5 мм) слой гранул, адсорбированных или иным образом закрепленных на поверхности пластинки из стекла или пластика, позволяет осуществлять Хроматографию в тонком слое, или "тонкослойную Хроматографию" (ТСХ). Английское обозначение TLC (thin layer chromatography). Движение жидкой фазы происходит за счет капиллярных сил.
Вместо тонкого слоя сорбента на основе целлюлозы можно использовать просто фильтровальную бумагу, иногда специальную - с введенными в нее ионогенными группами. Соответствующий процесс следует называть хроматографией на бумаге.
Вместо бумаги для аналогичного типа хроматографии используют пленки из модифицированной целлюлозы, полиамидные пленки и т.д. - это варианты хроматографии на пленках,
Пластинки, бумага или пленка могут располагаться горизонтально или вертикально; в последнем случае движение подвижной фазы может быть восходящим или нисходящим - это не играет принципиальной роли, так как оно обусловлено в основном капиллярными силами. Препараты на пластинки или бумагу чаще всего наносят в виде полоски или пятна раствора у одного края сорбента, неподалеку от уровня элюирующей жидкости, в которую этот край погружают. В последнее время для ТСХ все чаще применяют вариант кольцевой хроматографии, когда исходный препарат наносят в виде кольца, а элюция идет радиально.
Лекция 6. Материалы матриц сорбентов и обменников. Техника колоночной хроматографии
Матрицей называют твердую основу неподвижной хроматографической фазы. Она имеет вид сплошных или пористых гранул; последние часто представляют собой пространственную сетку линейных полимеров. Для придания материалу матрицы необходимых для хроматографии свойств его модифицируют. Модификация может представлять собой химическое присоединение ("присадку") ионогенных групп, гидрофобных молекул, биологически активных веществ или фиксацию путем адсорбции тонкого слоя растворителя. Хотя особенности хроматографического процесса определяются в основном характером модификации, физико-химические параметры матрицы могут существенно влиять на свойства неподвижной фазы. К таким параметрам относятся следующие: размеры и форма гранул и их пор; диапазон разброса этих размеров; механическая прочность материала матрицы; характер его смачивания и набухания в элюенте; химическая стойкость и инертность в условиях хроматографической элюции; реакционная способность, обеспечивающая возможность химической модификации матрицы.
Одни и те же матрицы, по-разному модифицированные, могут образовывать неподвижную фазу для разных хроматографических методов, поэтому во избежание повторений в данной главе мы познакомимся с физико-химическими свойствами всех применяемых в настоящее время матриц, а их модификации (а также свойства и номенклатуру получаемых путем этих модификаций сорбентов) рассмотрим в последующих главах, посвященных различным методам хроматографии. Прежде чем перейти к анализу свойств различных матриц, укажем общие для них способы обозначения диапазона линейных размеров гранул (а для сфер - их диаметров). Этот диапазон указывают либо непосредственно в микронах, либо в виде интервала чисел "МЕШ" ("mesh"). Число МЕШ соответствует числу нитей на дюйм в сетке с квадратными ячейками, через которую просеиваются гранулы. С учетом толщины самих нитей это означает, что через сетку в 100 МЕШ пройдут все гранулы, максимальный размер которых меньше 160 мкм, через сетку в 200 МЕШ - меньше 80, 400 МЕШ - меньше 40. Таким образом, диапазон размеров гранул 40-80 мкм соответствует 200-400 МЕШ. В него попадут гранулы, которые просеиваются через сетку в 200 МЕШ, но не проходят через ячейки сетки в 400 МЕШ. Для мелких гранул, размер которых менее 40 мкм, принято обозначение - 400 МЕШ.
ХРОМАТОГРАФИЧЕСКИЕ КОЛОНКИ.
Колонки, изготовленные в лаборатории.
В лабораторной стеклодувной мастерской несложно изготовить стеклянную колонку диаметром до 8 см и длиной до 1,5 м. Сверху колонка снабжена стандартным шлифом № 14 или. № 29, куда вставляется шлифованная пробка с капельницей. Последняя снабжена оливой малого диаметра, на которую можно надеть трубочку из силиконовой резины, куда вставляется тонкая (1-2 мм) полиэтиленовая или тефлоновая трубка от насоса. На нижний конец капельницы целесообразно надегь с некоторым перекосом кусочек полиэтиленовой трубочки так, чтобы при установке пробки на место трубочка почти касалась стенки колонки. Этим предотвращается взмучивание верхнего слоя сорбента при падении капли. Верхнюю половину поверхности шлифа смазывают силиконовой смазкой, но так, чтобы смазка не попадала внутрь колонки. В момент установки в гнездо пробку немного проворачивают. Если шлиф хорошо притерт, слой смазки должен быть прозрачным. Герметичность посадки пробки следует проверять перед каждым опытом - при неработающем насосе жидкость из колонки не должна вытекать. В более простом варианте шлифованную стеклянную пробку можно заменить на резиновую.
В нижнюю часть колонки заплавляется стеклянный фильтр. Чтобы не забиваться, его поры должны быть заведомо меньшего размера, чем гранулы сорбента. Вместе с тем нежелательно, чтобы фильтр представлял собой большое сопротивление току элюента. Для сорбентов, используемых при обычной хроматографии низкого давления, с гранулами не мельче 30 мкм в поперечнике подходит стеклянный фильтр № 3. Однако он может постепенно забиваться, если используется сорбент, засоренный "пылью", образующейся при его истирании. Во избежание этого не следует пренебрегать описанной ниже операцией "отмучивания" сорбента. Следует также помнить о том, что стеклянный фильтр сорбирует некоторое количество белка или нуклеиновой кислоты. С этой точки зрения в качестве фильтров следует предпочесть полиамидные (найлон) или тефлоновые пористые пластинки. Однако колонку в этом случае придется делать сборной, что значительно усложняет ее конструкцию. Такие фильтры используются в продажных фирменных колонках.
Под фильтром, при переходе к сливной трубке малого диаметра образуется коническая полость. При изготовлении колонки надо стремиться к тому, чтобы объем этой полости был минимальным, так как в ней может происходить смешивание близко идущих фракций. На сливной трубке имеется такая же, как на пробке, олива для трубочки из силиконовой резины, куда вставляется тонкая трубка, идущая к денситометру. Резиновую трубочку удобно пережимать винтовым зажимом, "запирая" таким образом выход из колонки.
Лекция 7. Адсорбционная и распределительная хроматографии
В адсорбционной хроматографии разделение веществ, входящих в смесь и движущихся по колонке в потоке растворителя, происходит за счёт их различной способности адсорбироваться и десорбироваться на поверхности адсорбента с развитой поверхностью, например, силикагеля.
В распределительной ВЭЖХ разделение происходит за счет разной растворимости разделяемых веществ в неподвижной фазе, как правило, химически привитой к поверхности неподвижного носителя, и подвижной фазе - растворителе. Этот метод разделения наиболее популярен, особенно в случае, когда привитая фаза представляет собой неполярный алкильный остаток от C8 до C18, а подвижная фаза более полярна, например смесь метанола или ацетонитрила с водой. Это так называемая обращённо-фазная (обратно-фазная, или с обращением фаз) хроматография.
Лекция 8. Тонкослойная хроматография
Тонкослойная хроматография (ТСХ) первоначально была разработана для разделения липидов. Хотя хроматография на бумаге быстрее, чем хроматография на колонке, к недостаткам ее следует отнести то, что бумага может быть изготовлена только из материалов на основе целлюлозы, что не позволяет применять ее для разделения неполярных веществ. Тонкослойная хроматография сохраняет все преимущества хроматографии на бумаге, но при этом позволяет использовать любой материал, который можно тонко измельчить и получить затем однородный слой. Это могут быть неорганические вещества, например силикагель, окись алюминия, диатомовая земля и силикат магния, а также органические вещества, в частности целлюлоза, полиамиды и порошок полиэтилена.
Пластинку с закрепленным сорбентом помещают в камеру, содержащую растворитель, и проявляют восходящей хроматографией. После того как фронт растворителя почти достигнет верхнего края, пластинку вынимают из камеры и сушат. Для получения двумерной хроматограммы высушенную пластинку можно повторно хроматографировать под прямым углом в другом растворителе. Положение пятен, как и при хроматографии на бумаге, определяют по окраске, по флуоресценции или при опрыскивании различными реагентами, которые реагируют с веществами в пятне с образованием окрашенных продуктов. Обычно используют следующие реагенты: нингидрин для аминокислот, родамин В для липидов, хлорид сурьмы для стероидов и терпенов, серную кислоту с последующим нагреванием практически для всех органических соединений (происходит обугливание), перманганат калия в серной кислоте для углеводородов, анисовый альдегид в серной кислоте для углеводов, пары брома для олефинов и т.д. Вещества можно элюировать путем соскребания.
Лекция 9. Ионообменная хроматография
ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ, жидкостная хроматография, основанная на разл. способности разделяемых ионов к ионному обмену с фиксир. ионами сорбента, образующимися в результате диссоциации ионогенных групп последнего. Для разделения катионов используют катиониты, для разделения анионов - аниониты (см. Иониты). Элюентом в первом случае служит р-р кислоты, во втором - р-р щелочи. Разделение ионов регулируют подбором оптим. значений рН элюента. Сильнокислотные сульфокатиониты и высокоосновные аниониты могут использоваться при любых значениях рН, слабокислотные карбоксильные катиониты - только при рН > 6; слабоосновные аниониты находятся в ионизованном состоянии при рН < 8. Варьируя рН элюента, можно резко изменять степень ионизации компонентов разделяемой смеси (сорбатов) и, следовательно, время их удерживания, добиваясь необходимой селективности разделения. Многозарядные ионы удерживаются ионитом сильнее однозарядных. При равных величинах зарядов удерживание падает с ростом радиуса гидратир. иона. Поэтому при И. х. в разб. р-рах на сульфокатионитах время удерживания катионов падает в ряду: Ва2+ > РЬ2+ > Sr2+ > Са2+ > Ni2+ > Cd2+ > Сu2+ > Со2+ > Zn2+ > Mg2+ > UO2+ > Тl+ > Ag+ > Cs+ > Rb+ > K+ > NH4+ > Na+ > H+ > Li+. Для орг. ионов на электростатич. взаимод. с фиксир. зарядами ионита накладывается еще и гидрофобное взаимод. орг. части иона с матрицей ионита. Чтобы уменьшить его вклад в удерживание орг. ионов и добиться оптим. селективности их разделения, к водному элюенту добавляют орг. компонент (1-25% метанола, изопропанола, ацетонитрила или диоксана). Элюент в И. х. кроме к-ты или основания и орг. добавок может содержать нейтральный электролит, напр. NaNO3, ионы к-рого конкурируют с разделяемыми ионами за взаимод. с сорбентом; при этом удерживание однозарядных ионов падает пропорционально концентрации соли в р-ре, двухзарядных ионов - пропорционально ее квадрату. Важна также природа нейтрального электролита: чем выше сродство его ионов к сорбенту, тем выше элюирующая сила р-ра. В И. х. анионов часто используют фосфатные р-ры, к-рые обладают большой элюирующей способностью при высоких значениях рН, где фосфат приобретает заряд - 3. Мн. неорг. катионы разделяют на сульфокатионитах, используя в качестве элюента комплексообразователи (орг. к-ты или гидроксикислоты). Разделение основано на том, что константы устойчивости образующихся комплексов, а значит и их сорбционные св-ва, даже таких близких по св-вам катионов, как лантаноиды и актиноиды, при определенных значениях рН различаются достаточно сильно; при этом заряд комплекса можно менять (вплоть до отрицательного). С помощью И. х. разделяют нек-рые нейтральные соед., если они способны превращ. в заряженные комплексы, как, напр., комплексы углеводов с борат-ионом. Удерживание разделяемых ионов в колонке пропорционально обменной емкости ионита. Для используемых в И. х. полимерных ионитов емкость 3-6 мг-экв/г, для ионитов на основе силикагеля с привитыми к его пов-сти функц. группами - на порядок ниже. При равном размере зерен (обычно 5-15 мкм) иониты на основе силикагеля обладают более высокой скоростью ионного обмена, что повышает эффективность хроматографич. колонок, однако их гидролитич. устойчивость при рН / 8 недостаточна. Для увеличения эффективности (числа теоретич. тарелок) колонки с полимерными ионитами обычно используют при повыш. т-рах (50-80 °С); при этом увеличиваются коэф. диффузии ионов в фазе ионита. В качестве сорбентов для И. х. могут использоваться нейтральные носители, пропитанные жидкими ионитами, т.е. несмешивающимися с водой орг. основаниями или к-тами, напр., триоктиламином, триоктилметиламмонием, алкиловыми зфирами алкилфосфорной к-ты. Разбавленные р-ры ионогенных ПАВ в сочетании с нейтральными гидрофобными носителями находят применение в ион-парной хроматографии (см. Жидкостная хроматография), к-рая отличается высокой эффективностью и большим числом варьируемых параметров для подбора оптим. селективности разделения. Детектирование в И. х. осуществляют с помощью любого детектора, применяемого в жидкостной хроматографии (см. Детекторы хроматографические). Наиб. универсален для ионных соединений кондуктометр, на применении к-рого основан вариант И. х. - ионная хроматография.И. х. применяется для разделения катионов металлов, напр., смесей лантаноидов и актиноидов, Zr и Hf Мо и W, Nb и Та; последние разделяют на анионитах в виде анионных хлоридных комплексов в р-рах соляной и плавиковой к-т. Щелочные металлы разделяют на катионитах в водных и водно-орг. средах, щел. - зем. и редкоземельные металлы на катионитах в присут. комплексонов. Большое значение имеет автоматич. анализ смесей прир. аминокислот на тонкодисперсном сульфокатионите в цитратном буфере при повыш. т-ре. Аминокислоты детектируют фотометрически после их р-ции с нингидрином или флюориметрически после дериватизации фталевым альдегидом. Высокоэффективная И. х. (колонки, упакованные сорбентом с размером зерен 5-10 мкм, давление для прокачивания элюента до 107 Па) смесей нуклеотидов, нуклеозидов, пурияовых и пиримидиновых оснований и их метаболитов в биол. жидкостях (плазма крови, моча, лимфа и др.) используется для диагностики заболеваний. Белки и нуклеиновые к-ты разделяют с помощью И. х. на гидрофильных высокопроницаемых ионитах на основе целлюлозы, декстранов, синтетич. полимеров, широкопористых силикагелей; гидрофильность матрицы ионита уменьшает неспецифич. взаимод. биополимера с сорбентом. В препаративных масштабах И. х. используют для выделения индивидуальных РЗЭ, алкалоидов, антибиотиков, ферментов, для переработки продуктов ядерных превращений.
Лекция 10. Ионообменная ЖХВД белков. Хроматофокусирование
Хроматофокусирование - метод ионообменной хроматографии, использующий в качестве инструмента разделения градиент рН, формируемый в слое сорбента внутри колонки. Метод успешно применяют для разделения цвиттер-ионных биологических макромолекул [1-3]. Формирование внутреннего градиента рН в хроматофокусировании заключается в предварительном уравновешивании анионообменной хроматографической колонки стартовым раствором (СР) с высоким значением рН, содержащим слабое основание, и в последующем пропускании элюента с более низким рН, составленного из слабых органических кислот или амфолитов.
Предложен вариант хроматофокусирования переходных металлов, сочетающий принципы комплексообразовательной хроматографии с формированием нисходящего градиента рН внутри колонки, заполненной сорбентом с привитыми олигоэтиленаминами.
В основе этого варианта лежит образование аминных комплексов ионов металлов (например, Cu2+, Co2+, Ni2+, Zn2+, Cd2+, Fe3+, Cr3+, Mn2+, UO22+, Pb2+) при начальном значении рН 7,0 - 7,5 и их последующая диссоциация при линейном снижении рН до 3. Возможности метода продемонстрированы на примере концентрирования и разделения 4 - 5 ионов металлов из изученного ряда в вариантах хроматографии низкого давления и ВЭЖХ [4, 5]. На данный момент ведутся работы на карбоксильных катионообменных сорбентах.
Это связано с тем, что комплексообразование ионов металлов с олигоэтиленаминами является многоступенчатым процессом с медленной кинетикой. В случае с карбоксильными сорбентами, вероятно, удастся расширить круг ионов металлов, разделяемых в условиях формирования нисходящего градиента рН, включив в него щелочноземельные металлы. Отметим, что нисходящие градиенты рН внутри катионообменных колонок в хроматофокусировании ранее не получали.
Лекция 11. Аффинная хроматография
АФФИННАЯ ХРОМАТОГРАФИЯ (от лат. affinis - родственный) (биоспецифич. хроматография, хроматография по сродству), метод очистки и разделения белков, основанный на их избират. взаимод. с лигандом, ковалентно связанным с инертным носителем. В кач-ве лигандов используют соед., взаимод. к-рых с разделяемыми в-вами основано на биол. ф-ции последних. Так, при разделении ферментов (для чего преим. и применяется А. х) лигандами служат их субстраты, ингибиторы или коферменты. Главная особенность, к-рая обусловливает высокую эффективность А. х., состоит в том, что разделение основано на различии не физ. - хим. признаков молекулы (заряда, формы и размера), а специфич. функциональных св-в, отличающих данный фермент от множества др. биополимеров.
Неподвижная фаза в А. х. представляет собой специально получаемый сорбент, построенный обычно по схеме: носитель - соединяющее звено ("ножка") - специфич. лиганд. Носителем служит чаще всего сефарозапроизводное агарозы, имеющее поперечные сшивки. Присоединение к ней лиганда или "ножки", содержащих, как правило, аминогруппу, осуществляется после активации сефарозы бромцианом:
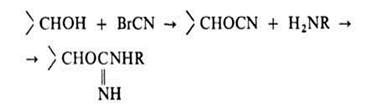
Содержание лиганда колеблется от 0,1 до 10 мкмоль на 1 г влажного сорбента. Сефароза, однако, малоустойчива к действию ряда хим. в-в и микроорганизмов.
Более стабильны макропористые неорг. носители (кремнезем,
стекло) и орг. полимеры. Если лиганд присоединяется непосредственно к носителю,
эффективность специфич. взаимод. с ферментом заметно снижается вследствие
пространств. затруднений. "Ножка", как правило, устраняет стерич. препятствия,
отдаляя лиганд от носителя. Как и носитель, она должна быть инертной и не
влиять на процессы в ходе А. х., чего, однако, не всегда удается достигнуть. Напр.,
присоединение "ножки" по приведенной выше р-ции приводит к
образованию катионной группировки изомочевины, и сорбент приобретает св-ва
анионита. В кач-ве "ножки" используют обычно ди - и полиамины,![]() аминокислоты, пептиды, олигосахариды.
аминокислоты, пептиды, олигосахариды.
Лигандами могут служить субстраты (напр., крахмал или гликоген при разделении амилаз), однако их превращ. в ходе А. х., катализируемое разделяемым ферментом, постоянно изменяет св-ва сорбента. Поэтому, как правило, применяют аналоги субстратов, устойчивые к дальнейшему превращ., т.е. ингибиторы ферментов. Так, для выделения протеиназ используют не расщепляемые ими пептиды D-аминокислот. Эффективны прир. ингибиторы ферментов, напр. пепстатин - ингибитор аспартильных протеиназ. Иногда применяют лиганды, связывающие большие группы родственных ферментов (в частности, киназы и дегидрогеназы). Примеры таких "группоспецифич." лигандов-антрахиноновые красители, аналоги никотинамидадениндину-клеотида.
Известны лиганды (напр., производные фенилборной к-ты), имитирующие при взаимод. с ферментом структуру переходного комплекса с субстратом. Такие лиганды эффективны при выделении сериновых гидролаз.
Разделение в А. х. обычно проводят на хроматографич. колонках; иногда разделяемую смесь помещают в сосуд с сорбентом и выдерживают до полного связывания исследуемого компонента. Затем сорбент (в колонке или сосуде) промывают буферным р-ром для удаления несвязавшихся в-в, после чего десорбируют исследуемый компонент.д.есорбция (элюция) последнего обычно достигается повышением ионной силы, изменением рН буферного р-ра или добавлением в него орг. р-рителя, что ослабляет взаимод. лиганд - фермент. Более избирательна десорбция р-ром лиганда.
Помимо ферментов, методом А. х. можно выделять также токсины, рецепторы, ингибиторы, транспортные белки и др. биологически активные в-ва. Высокой избирательностью отличается т. наз. иммуносорбция, при к-рой в кач-ве лиганда используют антитела, обладающие специфичностью к выделяемым белкам; особенно эффективны моноклональные антитела.
Для разделения белков применяется также ряд др. аналогичных методов.Т. наз. ковалентная хроматография основана на избират. образовании и последующем расщеплении ковалентных связей между выделяемым в-вом и носителем, напр. между белком с SH-группами и ртуть-орг. производными агарозы.
Применяется также лигандообменная хроматография, при к-рой ферменты связываются через функциональный ион металла с комплексоном, иммобилизованным на носителе.
Получила распространение гидрофобная хроматография, при к-рой сорбент (напр., фенилсефароза), содержащий гидрофобные группировки, вкрапленные в гидрофильную матрицу, взаимодействует с гидрофобными участками, содержащимися на пов-сти белков. Нередко при этом наблюдаются также ионообменные взаимод., как, напр., при использовании в качестве сорбента алкиламиносефароз. Избират. выделение гликопротеинов обеспечивают иммобилизованные на носителях лектины - белки, специфически взаимодействующие с концевыми моносахаридными звеньями углеводных цепей.
Иммобилизованные субъединицы ряда белков с четвертичной структурой м. б. использованы для извлечения этих белков из сложных смесей вследствие специфич. межсубъединичных контактов. А. х. сформировалась как метод в кон.60-х гг.20 в.
Лекция 12. Гель-фильтрация
ГЕЛЬ-ФИЛЬТРАЦИЯ ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДА.
Привычное название "гель-фильтрация" для хроматографического метода фракционирования молекул по их размерам можно сохранить и в случае хроматографии при высоком давлении, где среди используемых жестких пористых матриц главную роль играет силикагель. Иногда используют и такие названия, как "молекулярно-ситовая" ("molecular sieve") или "эксклюзивная" ("exclusion... ") хроматография.
Неподвижная фаза при гель-фильтрации представлена жидкостью, находящейся внутри пористых, хорошо смачиваемых гранул, заполняющих хроматографическую колонку. Если на такую колонку подается растворенная в элюенте смесь молекул различных размеров, то крупные молекулы, неспособные проникнуть внутрь гранул, будут двигаться вдоль колонки вместе с подвижной фазой;
для них коэффициент распределения К == 0. В то же время наиболее мелкие молекулы, размеры которых заведомо меньше диаметра пор в гранулах, будут равномерно распределяться между подвижной и неподвижной фазами. Для них будет осуществляться хроматографический процесс с присущим ему замедлением миграции хроматографической зоны; значение К при этом близко к единице. Для молекул промежуточной величины благодаря статистическому распределению размеров пор окажется доступной только часть объема неподвижной фазы. Для них 0 < К < 1, поэтому зона или зоны таких молекул будут мигрировать вдоль колонки быстрее, чем мелкие молекулы, но медленнее, чем крупные. В результате произойдет фракционирование исходной смеси молекул на зоны в зависимости от их размеров. Зоны выходят из колонки в порядке убывания этих размеров. В простейшем случае, когда в исходной смеси содержатся молекулы только двух категорий (крупные и мелкие), гель-фильтрация позволяет осуществить "сортировку" этих молекул ("group separation"). В частности, таким образом проводят обессоливание растворов биополимеров и очистку макромолекул от сопутствующих им низкомолекулярных компонентов, например от "предшественников", участвующих в их биосинтезе. Смесь молекул нескольких промежуточных размеров в ходе гель-фильтрации разделяется на ряд дискретных групп, различающихся между собой по степени доступности для них объема внутри гранул. Соответствующие хроматографические зоны мигрируют с различными скоростями и выходят из колонки в виде разделившихся "пиков".
Лекция 13. Теоретические и методические основы электрофореза
ЭЛЕКТРОМИГРАЦИОННЫЕ МЕТОДЫ, методы исследования в р-рах
ионизир. в-в и разделения их сложных смесей; основаны на явлении переноса
заряженных частиц в электрич. поле, приложенном к изучаемому р-ру. Осн. параметр,
характеризующий перенос частиц, - подвижность и, т.е. расстояние l,
нак-рое в-во переместится под действием единицы градиента электрич. потенциала Е
за единицу времени![]()
![]()
Процессы, связанные с комплексообразованием, ассоциацией или пересольватацией ионов, а также с изменением состояния р-рителя, приводят к изменению заряда либо радиуса ионов, что оказывает влияние на их подвижность; на этом основано применение Э. м. для исследования р-ций в р-рах.
Неодинаковая подвижность мол. ионов и заряженных частиц разл. хим. природы позволяет использовать Э. м. также для разделения смесей; в данном случае эти методы часто наз. электрофорезом.
Методы измерения подвижности заряженных частиц. Подвижность, или скорость миграции индивидуальных ионов, можно определять:
1) по изменению концентрации ионов исследуемого элемента в приэлектродном пространстве при электролизе;
2) путем смещения в электрич. поле узких зон изучаемых ионов;
3) с помощью подвижной границы между зонами (фронтальные методы, изотахофорез).
Исследование реакций в растворах. Информацию о равновесных процессах в р-ре получают при изучении зависимости скорости миграции ионов исследуемого элемента от концентрации одного или неск. участвующих в р-ции в-в. По этой зависимости можно выявлять состав продуктов р-ции и определять константы равновесия.
В случае р-ций комплексообразования изучаемый металл М может находиться одновременно в неск. ионных формах связи с лигандом А, между к-рыми устанавливается подвижное равновесие. В такой системе общее, или суммарное, перемещение в электрич. поле всех ионов, содержащих М и имеющих индивидуальные подвижности иi, происходит с нек-рой ср. скоростью ис, характеризующей суммарный электромиграц. перенос металла в единицу времени:
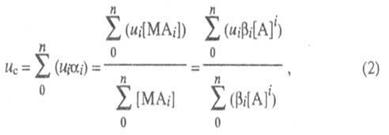
где i - число лигандов в комплексе; ![]() - доля
металла, связанного в i-ую ионную форму;
- доля
металла, связанного в i-ую ионную форму; ![]() - полная
константа устойчивости ионной формы; [М], [А] и [МАi] - соотв.
равновесные концентрации металла, лиганда и комплекса.
- полная
константа устойчивости ионной формы; [М], [А] и [МАi] - соотв.
равновесные концентрации металла, лиганда и комплекса.
Кривая электромиграции (рис.1), отражающая смещение подвижного равновесия между разл. ионными формами при изменении равновесной концентрации лиганда, устанавливает области существования: своб. ионов (I); координационно ненасыщенных форм (II); координационно насыщенных комплексных ионов (III).
Состав комплексных ионов можно определять неск. приемами: по эмпи-рич. зависимости между подвижностью ионов и величиной их заряда; из соотношения общей и равновесной концентраций лиганда, к-рое определяется по скорости электромиграции введенного в систему вспомогат. металла (по ур-нию 2); по соотношению между коэф. диффузии и подвижностью при одной и той же концентрации лиганда. Константы устойчивости ионных форм рассчитывают путем решения системы из п ур-ний вида (2), где п равно числу ионных форм.
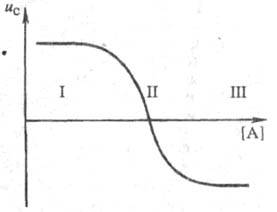
Рис.1. Зависимость средней скорости миграции металла ис от концентрации лиганда [А]: I, II, Ш - области осуществления соотв. своб. ионов металла, координационно ненасыщенных форм и координационно насыщенных комплексных ионов.
При исследовании хим. взаимодействий в р-ре важно сохранение постоянства состава фонового электролита, к-рое может нарушаться вследствие электродных р-ций. Поэтому целесообразно использовать аппаратуру, предотвращающую проникновение продуктов электролиза в рабочую часть прибора. Если это условие выполнено, то Э. м. дают достаточно правильные результаты благодаря тому, что отсутствует необходимость введения в изучаемую систему новых фаз - ионообменных смол, экстрагентов и т.п., вызывающих побочные равновесные процессы.
Помимо равновесий Э. м. позволяют исследовать кинетику комплексообразования, используя явление дополнит. квазидиффузионного размывания под действием электрич. поля первоначально узкой зоны, содержащей разл. ионные формы с разными знаками заряда. Для достижения этого эффекта необходимо приложить поле такой напряженности, чтобы скорость электромиграц. переноса ионов превышала скорость протекания комплексообразования.
Для изучения кинетики р-ций можно использовать также зонную электромиграцию ионов в неравновесном с ними электролите. В обоих случаях применение Э. м. целесообразно при анализе процессов, скорость к-рых лежит в пограничной области между медленными и быстрыми р-циями.
Закономерности электромиграции ионов в расплавах солей исследованы в значительно меньшей степени, чем в водных р-рах. Обычная среда при проведении электромиграции в расплавленных солях - безводные расплавы нитратов и перхлоратов щелочных металлов или эвтектич. смеси, имеющие сравнительно низкую т-ру плавления.
Для электромиграции в расплавах характерны две осн. проблемы: сильное взаимод. ионов изучаемых металлов и расплавленной соли; отсутствие электрически нейтрального р-рителя, для к-рого можно измерить истинную скорость движения ионов. Поэтому обычно их подвижность определяют относительно прибора, в к-ром проводят электромиграцию. В этом случае данные о подвижности в расплавах смещены на неизвестную постоянную величину.
Лекция 14. Изоэлектрическое фокусирование и изотахофорез
Фокусирующий ионный обмен. Этот метод часто наз. электрофоретич. фокусировкой или просто электрофокусированием, связан с наложением градиента концентрации или рН р-ра параллельно электрич. полю. Благодаря этому разделяемые ионы могут изменять величину и знак заряда по мере перемещения в поле градиента. При этом в фиксир. точках системы каждый компонент переходит в изоэлектрич. состояние, в к-ром ср. заряд частиц данного компонента равен нулю. Упомянутые точки являются местом концентрирования (фокусирования) отдельных компонентов смеси (рис.5). Положение зон фокусирования определяется градиентом концентрации комплексообразующего реагента или рН р-ра и константами устойчивости комплексных ионов разделяемых элементов. При разделении смеси белков или др. амфотерных соед. положение зон определяется значениями их изоэлектрич. точек.
Для создания градиента рН электродные камеры заполняются буферными р-рами с разными значениями рН. Напр., для разделения редкоземельных элементов цериевой группы в 0,001 М р-ре этилендиаминтетрауксусной к-ты рН должен изменяться по длине колонки от 1,7 у анода до 2,4 у катода.
В сер.60-х гг.20 в. было предложено создавать градиент рН с помощью амфолитов - смесей алифатич. полиаминокислот. Под влиянием электрич. поля амфолиты распределяются в соответствии со своими изоэлекгрич. точками и тем самым образуют градиент рН. Применение амфолитов позволяет добиться весьма высокой разрешающей способности метода: в нек-рых случаях удается разделить белки, изоэлектрич. точки к-рых различаются на 0,02 единицы рН.
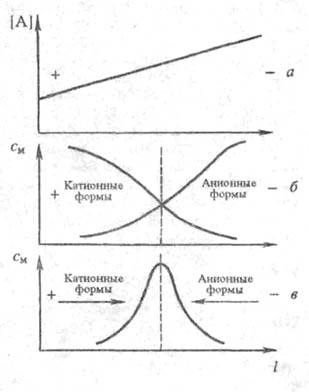
Схема, поясняющая метод электрофокусирования: а - образование градиента концентрации лиганда [А] ; б - распределение ионных форм мигранта; в - концентрирование лиганда в узкой зоне; см - концентрация мигранта.
Описанный метод, являясь самостоятельным, в то же время представляет собой вариант зонного электрофореза. Во всех модификациях последнего идентификацию и количеств. определение в-в в зонах можно проводить как непосредственно на носителе, так и после элюирования. В обоих случаях используют методы радиоактивных индикаторов, фотометрию в прямом и отраженном свете, люминесцентный анализ.
Фронтальные методы основаны на измерении скорости перемещения границы раздела р-ров с разной плотностью. Классич. вариант метода был разработан в 1930 и с тех пор применяется для определения подвижности и разделения высокомол. в-в, в частности белков. В простейшей модификации метода в U-образную трубку помещают р-р белков, а над ним буферный электролит, в к-рый погружены электроды. При наложении электрич. поля индивидуальные белки перемещаются с разл. скоростями, образуя серию границ. Их положение регистрируют оптич. методами по изменению коэф. преломления.
Изотахофорез. Осн. частью прибора служит капиллярная трубка с анодным и катодным резервуарами на концах. При анализе анионов анодное отделение и капилляр заполняют т. наз. лидирующим электролитом, содержащим анион с высокой подвижностью. Ср. скорость миграции анионов в этом электролите должна быть выше подвижности любого аниона в исследуемой смеси. Катодное отделение заполняют т. наз. замыкающим электролитом, анион к-рого имеет подвижность меньшую, чем подвижность любого др. аниона в смеси. Анализируемый образец, в к-ром нужно определить содержание анионов, вносят между предшествующим и замыкающим электролитами. После подачи напряжения (5-10 кВ) при силе тока до 100 мкА по мере движения анионов к катоду постепенно образуются зоны индивидуальных анионов определенной длины, разделенные четкими границами, ширина к-рых составляет 0,2-0,3 мм при диаметре капилляра 0,1 мм. После этого все зоны будут перемещаться с одинаковой скоростью (отсюда назв. метода). Соотношение концентраций анионов в двух соседних зонах с1 и с2 в установившемся режиме будет определяться выражением Кольрауша:
с1/с2 = n1/n2, (4)
где n1и n2 - числа переноса.
При анализе катионов лидирующий электролит должен содержать катионы с высокой подвижностью, замыкающий - с миним. для данной системы скоростью миграции.
Кол-во в-ва в зоне Q и ее длина l в капилляре постоянного сечения S связаны простым соотношением:
Q = ClS, (5)
где С - коэф. пропорциональности.
В установившеся режиме градиент потенциала при переходе от лидирующего к замыкающему электролитам скачкообразно возрастает в соответствии с подвижностью ионов, составляющих данную зону. Это приводит к температурным скачкам между зонами, регистрируя к-рые с помощью термопары можно определить расстояние между зонами и по выражению (5) найти кол-во в-ва в зоне.
Лекция 15. Обнаружение, количественное определение и характеристика макромолекул после электрофореза
На электрофоретически разделенные антигены наносят иммунные сыворотки, содержащие различные специфические антитела. При встрече соответствующих антигена и антитела в зоне оптимального их соотношения наблюдается реакция преципитации невооруженным глазом. Иммунный электрофорез объединяет преимущества электрофореза и иммунной реакции: высокая разрешающая способность метода, разделяющая компоненты анализируемой системы на основе электрофоретической мобильности и высокая специфичность иммунных антисывороток.
Лекция 16. Принцип иммунного электрофореза. Иммунофиксация
Электрофорез с иммунофиксацией (JFE) - это двухступенчатый процесс, использующий электрофорез протеинов на первом этапе и иммунопреципитацию на втором. При этом исследованию может быть подвергнута сыворотка крови, моча, спинномозговая или другая жидкость организма. Электрофорез с иммунофиксацией - один из современнейших методов в кинической лаборатории для получения характеристик моноклональных иммуноглобулинов. Моноклональная гаммопатия характеризуется неконтролируемой пролифирацией одного клона плазменных клеток за счет других клеток. Эта дисфункция часто приводит к синтезу большого количества одного иммуноглобулина или его субъединицы со снижением нормальных уровней иммуноглобулинов. При этом на электрофореграмме выявляется один резко увеличенный пик в бета-гамма-области.
Лекция 17. Электросинерез. Электроиммуноанализ
Перекрестный иммуноэлектрофорез.
Информацию о равновесных процессах в р-ре получают при изучении зависимости скорости миграции ионов исследуемого элемента от концентрации одного или неск. участвующих в р-ции в-в. По этой зависимости можно выявлять состав продуктов р-ции и определять константы равновесия. В случае р-ций комплексообразования изучаемый металл М может находиться одновременно в неск. ионных формах связи с лигандом А, между к-рыми устанавливается подвижное равновесие. В такой системе общее, или суммарное, перемещение в электрич. поле всех ионов, содержащих М и имеющих индивидуальные подвижности иi, происходит с нек-рой ср. скоростью ис, характеризующей суммарный электромиграц. перенос металла в единицу времени:
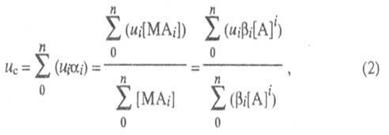
где i - число лигандов в комплексе;
![]()
- доля металла, связанного в i-ую ионную форму;
![]()
- полная константа устойчивости ионной формы; [М], [А] и [МАi] - соотв. равновесные концентрации металла, лиганда и комплекса.
Кривая электромиграции (рис.1), отражающая смещение подвижного равновесия между разл. ионными формами при изменении равновесной концентрации лиганда, устанавливает области существования: своб. ионов (I); координационно ненасыщенных форм (II); координационно насыщенных комплексных ионов (III).
Состав комплексных ионов можно определять неск. приемами: по эмпи-рич. зависимости между подвижностью ионов и величиной их заряда; из соотношения общей и равновесной концентраций лиганда, к-рое определяется по скорости электромиграции введенного в систему вспомогат. металла (по ур-нию 2); по соотношению между коэф. диффузии и подвижностью при одной и той же концентрации лиганда.
Константы устойчивости ионных форм рассчитывают путем решения системы из п ур-ний вида (2), где п равно числу ионных форм.
Лекция 18. Методы меченых атомов
ИЗОТОПНЫЕ ИНДИКАТОРЫ, в-ва, имеющие в своем составе хим. элемент с изотопным составом, отличающимся от природного. Часто И. и. называют сами изотопы-метки, добавляемые в в-во, содержащее прир. смесь изотопов данного элемента. Т.к. поведение изотопов одного элемента в физ. - хим. процессах практически идентично (за исключением легких элементов с атомными номерами Z = 10-12, для которых относительно большую роль могут играть изотопные эффекты), использование И. и. позволяет по регистрации изотопа-метки исследовать самодиффузию и миграцию меченого в-ва, определять ничтожно малые кол-ва в-ва, изучать механизмы хим. р-ций и биол. процессов (т. наз. метод изотопных индикаторов, ранее наз. методом меченых атомов).
Лекция 19. Спектрофотометрические методы анализа
СПЕКТРОФОТОМЕТРИЯ, метод исследования и анализа в-в, основанный на измерении спектров поглощения в оптич. области электромагн. излучения. Иногда под С. понимают раздел физики, объединяющий спектроскопию (как науку о спектрах электромагн. излучения), фотометрию и спектрометрию [как теорию и практику измерения соотв. интенсивности и длины волны (или частоты) электромагн. излучения] ; на практике С. часто отождествляют с оптич. спектроскопией. По типам изучаемых систем С. обычно делят на молекулярную и атомную. Различают С. в ИК, видимой и УФ областях спектра (см. Инфракрасная спектроскопия, Ультрафиолетовая спектроскопия).
Применение С. в УФ и видимой областях спектра основано на поглощении электромагн. излучения соединениями, содержащими хромофорные (напр., С = С, С=С, С=О) и ауксохромные (ОСН3, ОН, NH2 и др.) группы (см. Цветность органических соединений}. Поглощение излучения в этих областях связано с возбуждением электронов s-, p-и n-орбиталей осн. состояния и переходами молекул в возбужденные состояния: s: s*, n: s*, p: p* и n: p* (переходы перечислены в порядке уменьшения энергии, необходимой для их осуществления; см. также Молекулярные спектры). Переходы s: s* находятся в далекой УФ области, напр. у парафинов при ~ 120 нм. Переходы n: s* наблюдаются в УФ области; напр., орг. соед., содержащие n-электроны, локализованные на орбиталях атомов О, N, Hal, S, имеют Полосы поглощения при длине волны ок. 200 нм. Линии, соответствующие переходам p: p*, напр., в спектрах гетероциклич. соединений проявляются в области ок.250-300 нм и имеют большую интенсивность. Полосы поглощения, соответствующие переходам n: p*, находятся в ближней УФ и видимой областях спектра; они характерны для соед., в молекулах к-рых имеются такие хромофорные группы, как С = О, C = S, N = N. Так, насыщ. альдегиды и кетоны имеют максимумы поглощения при длине волны ок.285 нм. Переходы типа n: p* часто оказываются запрещенными, и соответствующие полосы поглощения обладают очень малой интенсивностью.
Переходы типа p: p* могут сопровождаться переходом электрона с орбитали, локализованной гл. обр. на одной группе (напр., С=С), на орбиталь, локализованную на др. группе (напр., С=О). Такие переходы сопровождаются переносом электрона с одного атома на другой и соответствующие спектры наз. спектрами с переносом заряда. Последние характерны для разл. комплексов (напр., арома-тич. соединений с галогенами), интенсивно поглощающих в видимой и УФ областях.
Для ионов переходных металлов и их комплексных соед. характерны переходы с участием d-электронов, а для РЗЭ и актиноидов-переходы с участием f-электронов. Соответствующие соед. в р-ре бывают интенсивно окрашенными, причем окраска (спектр поглощения) зависит от степени окисления катиона и устойчивости комплексного соединения. Поэтому С. широко используют при исследовании и анализе комплексных соед. металлов.
Изолированные, не взаимодействующие между собой хромофоры в молекуле поглощают независимо. В случае к. - л. взаимод. между ними аддитивность спектров нарушается. По отклонениям от аддитивности можно судить о характере и величине взаимодействия. Поскольку положение полос в спектре определяется как разность энергий основного и возбужденного состояний молекул, можно определять структуру энергетич. уровней молекул или по известной схеме энергетич. уровней определять положение полос поглощения. Любому электронному состоянию молекул соответствует набор разл. колебат. уровней энергии. Колебат. структура полосы, соответствующей переходу между электронными уровнями, может отчетливо проявляться не только в спектрах газов, но и в спектрах нек-рых р-ров, что дает возможность получать дополнит. информацию о взаимод. молекул. Спектрофотометрич. исследование спектров молекул в видимой и УФ областях позволяет установить вид электронных переходов и структуру молекул. При этом часто исследуют влияние разл. типов замещения в молекулах, изменения р-рителей, т-ры и др. физ. - хим. факторов.
В ИК области проявляются переходы между колебат. и вращат. уровнями (см. Колебательные спектры, Вращательные спектры). Среди частот колебаний молекул выделяют т. наз. характеристические, к-рые практически постоянны по величине и всегда проявляются в спектрах хим. соед., содержащих определенные функц. группы (вследствие чего эти частоты иногда называют групповыми; см. табл. на форзаце 2-го тома). Теория колебаний сложных молекул позволяет расчетным путем предсказать колебат. спектр соединений, т.е. определить частоты и интенсивности полос поглощения.
Колебат. спектры молекул чувствительны не только к изменению состава и структуры (т.е. симметрии) молекул, но и к изменению разл. физ. и хим. факторов, напр. изменению агрегатного состояния в-ва, т-ры, природы р-рителя, концентрации исследуемого в-ва в р-ре, разл. взаимод. между молекулами в-ва (ассоциация, полимеризация, образование водородной связи, комплексных соед., адсорбция и т.п.). Поэтому ИК спектры широко используют для исследования, качеств. и количеств. анализа разнообразных в-в.
В ближней ИК области (10000-4000 см-1, или 1-2,5 мкм), где расположены обертоны и составные частоты осн. колебаний молекул, полосы поглощения имеют интенсивность в 102-103 раз меньше, чем в средней ИК области (4000-200 см-1). Это упрощает подготовку образцов, т.к толщина поглощающего слоя м. б. достаточно большой (до неск. мм и более). Эксперим. техника для работы в этой области относительно проста. Однако чувствительность и селективность определения отдельных соед. невелики. Тем не менее высокое отношение сигнал: шум (до 105) создает хорошие условия для количеств. анализа при содержании определяемого соед. ок.1% и выше. Подобные анализы выполняются за 1 мин. В дальней ИК области (200-5 см-1) могут наблюдаться чисто вращат переходы.
Интенсивность полосы поглощения молекулы определяется вероятностью соответствующего электронного (или колебательного) перехода. Для характеристики интенсивности полосы служит молярный коэф. поглощения e (см. Абсорбционная спектроскопия), определяемый, согласно закону Бугера-Ламберта-Бера, как e = A/Cl, где А = = - lgT= - lg (I/I0), T-пропускание, I0 и I-интенсивности соотв. падающего и прошедшего через в-во излучения, С-молярная концентрация в-ва, поглощающего излучение, l-толщина поглощающего слоя (кюветы), в см. Обычно e<105, в ИК области e<2·103 (л/моль·см). Закон Бугера-Ламберта-Бера лежит в основе количеств. анализа по спектрам поглощения.
Для измерения спектров используют спектральные приборы-спектрофотометры, осн. части к-рого: источник излучения, диспергирующий элемент, кювета с исследуемым в-вом, регистрирующее устройство. В качестве источников излучения применяют дейтериевую (или водородную) лампу (в УФ области) и вольфрамовую лампу накаливания или галогенную лампу (в видимой и ближней ИК областях). Приемниками излучения служат фотоэлектронные умножители (ФЭУ) и фотоэлементы (фоторезисторы на основе PbS). Диспергирующими элементами прибора являются призмен-ный монохроматор или монохроматор с дифракц. решетками. Спектр получают в графич. форме, а в приборах со встроенной мини-ЭВМ-в графической и цифровой формах. Графически спектр регистрируют в координатах: длина волны (нм) и (или) волновое число (см-1) - пропускание (%) и (или) оптич. плотность. Осн. характеристики спектрофотометров: точность определения длины волны излучения и величины пропускания, разрешающая способность и светосила, время сканирования спектра. Мини-ЭВМ (или микропроцессоры) осуществляют автоматизир. управление прибором и разл. мат. обработку получаемых эксперим. данных: статистич. обработку результатов измерений, логарифмирование величины пропускания, многократное дифференцирование спектра, интегрирование спектра по разл. программам, разделение перекрывающихся полос, расчет концентраций отдельных компонентов и т.п. Спектрофотометры обычно снабжаются набором приставок для получения спектров отражения, работы с образцами при низких и высоких т-рах, для измерения характеристик источников и приемников излучения и т.п.
Для исследования спектров в ИК области используют обычно спектрофотометры, работающие в интервале от 1,0 до 50 мкм (от 10000 до 200 см-1). Осн. источниками излучения в них являются стержень из кароида кремния (глобар), штифт из смеси оксидов циркония, тория и иттрия (штифт Нернста) и спираль из нихрома. Приемниками излучения служат термопары (термоэлементы), болометры, разл. модели оптико-акустич. приборов и пироэлектрич. детекторы, напр. на основе дейтерированного триглицинсульфата (ТГС). В спектрофотометрах, сконструированных по "клас-сич." схеме, в качестве диспергирующих элементов применяют призменный монохроматор или монохроматор с дифракц. решетками. С кон.60-х гг.20 в. выпускаются ИК фурье-спектрофотометры (см. Фурье-спектроскопия), к-рые обладают уникальными характеристиками: разрешающая способность-до 0,001 см-1, точность определения волнового числа v-до 10-4 см-1 (относит. точность bDv/v!! 10 - 8), время сканирования спектра может достигать 1 с, отношение сигнал: шум превышает 105. Эти приборы позволяют изучать образцы массой менее 1 нг. К ним также имеются разл. приставки для получения спектров отражения, исследования газов при малых или высоких давлениях, разных т-рах и т.п. Встроенная в прибор мини-ЭВМ управляет прибором, выполняет фурье-преобразования, осуществляет накопление спектров, проводит разл. обработку получаемой информации.
Лекция 20. Флюориметрические методы анализа
Наиб. распространение получил анализ, основанный на фотолюминесценции исследуемого в-ва, возбуждаемой УФ излучением. Источниками последнего служат кварцевые газоразрядные ртутные или ксеноновые лампы и УФ лазеры. Pегистрируют люминесценцию визуально, фотографически или фотоэлектрически с помощью спектрографов, фотометров и спектрофотометров Л. а. подразделяют на качественный и количественный. Качеств Л. а. проводят по спектрам люминесценции. Его используют, напр., для обнаружения битумов в породах, следов люминесцирующих орг. и неорг. в-в в разл. объектах. Разновидность качеств. Л. а. - сортовой анализ, к-рый позволяет обнаруживать невидимые при обычном освещении различия в исследуемых объектах и используется для установления сортности и качества стекол, семян, с. - х. продукции, для определения минералов в породах, поверхностных и сквозных дефектов, выявления подделок, в криминалистике и т.д. Количеств Л. а. основан на зависимости интенсивности люминесценции от кол-ва люминесцирующего в-ва. Различают флуоресцентный, фосфоресцентный и хемилюминесцентный анализы. Флуоресцентный анализ основан на образовании люминесцирующих комплексных соед. элементов с орг. реагентами, напр. гидроксипроизводными флавона (морин, кверцетин), производными тригидроксифлуорона и гидроксиантрахинона, 8-оксихинолином, родаминами и др. Этот метод мало селективен, большинство реагентов - групповые, лишь люмогаллион специфичен для определения Ga и люмомагнезон - Mg. Для увеличения селективности используют экстракционно-флуоресцентный анализ - предварит. разделение анализируемой смеси методом экстракции, а также охлаждение р-ров до азотных и гелиевых т-р. В последнем случае может возникнуть фосфоресценция. Фосфоресцентный анализ обладает большой селективностью, т.к лишь немногие катионы образуют с орг. реагентами фосфоресцирующие комплексы, сами же реагенты не фосфоресцируют.д.ля регистрации спектров и интенсивности фосфоресценции используют фосфороскоп; при этом флуоресценция не регистрируется. Хемилюминесцентный анализ основан на свечении, возникающем в результате окислит. - восстановит. р-ций орг. в-в, напр. люминола, люцигенина и др., с катионами переходных металлов, напр. Fe (III), Co (II), Cu (II), Ni (II), Mn (II) и др.; концентрацию последних определяют по изменению интенсивности свечения. Предел обнаружения 5.10-7%. По собственной люминесценции определяют U, лантаноиды и нек-рые переходные элементы с большой селективностью, т.к их спектры в ряде случаев характеризуются структурой. Пределы обнаружения U в водах и геол. объектах при применении кристаллофосфоров 5.10-7 - 1.10-8%; РЗЭ при использовании орг. реагентов 103 - 10-4%, в кристаллофосфорах 10-5-10-6%; переходных элементов (в т. ч. и платиновых) в кристаллофосфорах 10-5-10-6%. Ртутеподобные ионы (Tl+, Pb2+, Bi3+, Те4+, As3+, Sb3+) можно определять по люминесценции замороженных р-ров их солей или в кристаллофосфорах с пределом обнаружения 10-4-10-7%. Применение лазеров позволяет снизить пределы обнаружения нек-рых элементов до 10-13%.Л. а. орг. соед. затруднен, т.к их спектры люминесценции, как правило, неспецифичны. Однако предложены методы количеств. определения порфиринов, витаминов, антибиотиков, хлорофилла и др. в-в, в спектрах к-рых имеются характеристичные полосы. При использовании лазеров пределы обнаружения достигают 10-7-10-11%. Ароматич. соед. в замороженных р-рах алифатич. углеводородов при т-рах 77 К дают характерные для каждого соед. квазилинейчатые спектры люминесценции (эффект Шпольского). Этот метод используют для определения полициклич. ароматич. углеводородов в экстрактах растений, почв, продуктов питания, горных пород и т.д. с пределом обнаружения 10-7-10-8%, а также для определения бензола, его гомологов и производных, ароматич. аминокислот при т-рах жидкого воздуха, азота, гелия в водно-солевой матрице с пределом обнаружения 10-4-10-6%.Л. а. используют в иммунохим. анализе для определения антител, гормонов, лек. препаратов, вирусных и бактериальных антигенов по концентрации комплекса антиген - антитело. При этом в иммунном флуоресцентном анализе к антителу непосредственно присоединяют флуоресцирующие в-ва, напр. РЗЭ, флуоресцирующие красители (чувствительность метода 10-14 моль/л), а в иммуноферментном анализе к антителу присоединяют фермент и в результате ферментативной р-ции, сопровождаемой биолюминесценцией, определяют ферментативную активность.
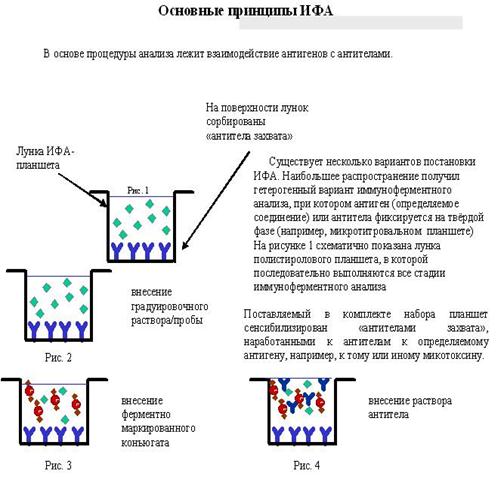
Лекция 21. Иммуноферментный анализ
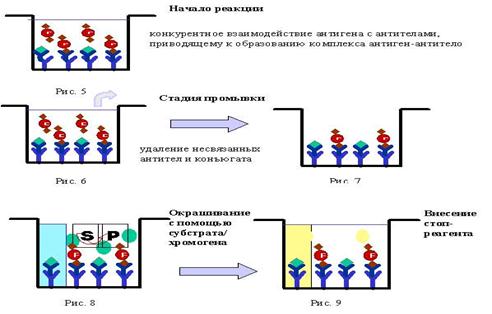
Лекция 22. Радиометрический анализ. Масс-спектроскопия
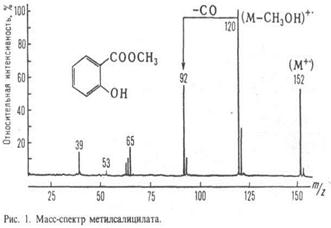
Метод анализа в-ва путем определения массы (чаще, отношения массы к заряду m/z) и относит. кол-ва ионов, получаемых при ионизации исследуемого в-ва или уже присутствующих в изучаемой смеси. Совокупность значений m/z и относит. величин токов этих ионов, представленная в виде графика или таблицы, наз. масс-спектром в-ва (рис.1).
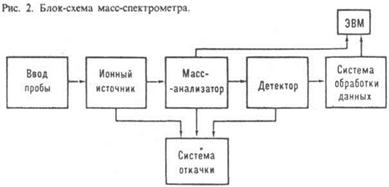
Начало развитию М. - с. положено опытами Дж. Томсона (1910), исследовавшего пучки заряженных частиц, разделение к-рых по массам производилось с помощью электрич. и магн. полей, а спектр регистрировался на фотопластинки. Первый масс-спектрометр построен А. Демпстером в 1918, а первый масс-спектрограф создал Ф. Астон в 1919; он же исследовал изотопич. состав большого числа элементов. Первый серийный масс-спектрометр создан А. Ниром в 1940; его работы положили начало изотопной М. - с. Прямое соединение масс-спектрометра с газо-жидкостным хроматографом (1959) дало возможность анализировать сложные смеси летучих соед., а соединение с жидкостным хроматографом с помощью термораспылит. устройства (1983) - смеси труднолетучих соединений. Macс-спектральные приборы. Для разделения ионов исследуемого в-ва по величинам m/z, измерения этих величин и токов разделенных ионов используют масс-спектральные приборы. Приборы, в к-рых регистрация осуществляется электрич. методами, наз. масс-спектрометрами, а приборы с регистрацией ионов на фотопластинках - масс-спектрографами. Масс-спектральные приборы состоят из системы ввода пробы (система напуска), ионного источника, разделительного устройства (масс-анализатора), детектора (приемника ионов), вакуумных насосов, обеспечивающих достаточно глубокий вакуум во всей вакуумной системе прибора, и системы управления и обработки данных (рис.2). Иногда приборы соединяют с ЭВМ:
Масс-спектральные приборы характеризуются чувствительностью, к-рая определяется как отношение числа зарегистрированных ионов к числу атомов введенной пробы. За абс. порог чувствительности принимают миним. кол-во исследуемого в-ва (выраженное в г, молях), за относительный - миним. массовую или объемную долю в-ва (выраженную в%), к-рые обеспечивают регистрацию выходного сигнала при отношении сигнал-шум 1: 1. Ионный источник предназначен для образования газообразных ионов исследуемого в-ва и формирования ионного пучка, к-рый направляется далее в масс-анализатор. наиб. универсальный метод ионизации в-ва - электронный удар. Впервые осуществлен П. Ленардом (1902). Совр. источники такого типа построены по принципу источника А. Нира (рис.3).
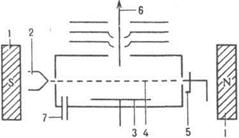
Рис.3. Схема ионного источника типа источника А. Нира: 1 - постоянный магнит; 2 - катод; 3 - выталкивающий электрод; 4 - поток электронов; 5 - ловушка электронов; 6 - ионный луч; 7 - ввод в-ва.
Для ионизации молекул обычно используют электроны с энергиями 70-100 эВ, к-рые движутся со скоростью 108 см/с и проходят путь, равный диаметру молекулы орг. соед. за 1016 с. Этого времени достаточно для удаления электрона из молекулы в-ва и образования мол. иона - положительно заряженного ион-радикала М+', имеющего энергию 2-8 эВ. Ионы с миним. запасом энергии достаточно устойчивы и достигают приемника. Ионы с большим запасом внутр. энергии распадаются на пути движения на ионы с меньшей мол. массой (т. наз. осколочные ионы), характерные для в-ва определенного строения. Для ионизации молекул энергия электронного пучка должна превышать нек-рую критическую для в-ва величину, наз. потенциалом ионизации. Потенциалы ионизации лежат в пределах 3,98 эВ (Fr) - 24,58 эВ (Не), для большинства орг. соед.7-11 эВ. Используя моноэнергетич. пучки электронов и снижая их энергию до пороговых значений, можно определять потенциалы ионизации в-в и потенциалы появления ионов - критич. энергию электронов, при к-рой в спектре появляются линии соответствующих осколочных ионов. При ионизации электронным ударом происходит перераспределение энергии возбуждения по колебат. степеням свободы мол. иона, прежде чем этот ион распадается. Предположение о квазиравновесном распределении энергии возбуждения позволяет полуэмпирич. путем рассчитать масс-спектры нек-рых в-в, согласующиеся с эксперим. данными. Однако во мн. случаях, особенно для длинных молекул, эта теория не подтверждается. Для двухатомных молекул изменения колебат. состояний объясняются, исходя из принципа Франка - Кондона (см. Квантовые переходы). При взаимод. низкоэнсргетич. электронов (менее 10 эВ) с в-вом могут осуществляться процессы резонансного захвата электронов молекулами с образованием отрицательно заряженных ионов М (см. также Ионы в газах).М. - с. электронного удара - высокочувствит. метод анализа, позволяет анализировать пикомольные кол-ва в-ва, ее предпочитают для исследования структуры соединений. Существуют "библиотеки" масс-спектров, содержащие спектры более 70000 орг. соед., по к-рым можно проводить их идентификацию с применением ЭВМ. Недостатки метода: мол. ионы образуются лищь у 20% орг. соед.; метод применим только для определения легколетучих термически стабильных соед.; в значениях полного ионного тока на ионы с большими значениями m/z, дающие информацию о мол. массе и наличии функц. групп, приходится меньшая часть; отрицательно заряженные ионы, имеющие большое значение в структурном анализе, образуются в очень небольшом кол-ве и ограниченным числом орг. соединений. Хим. ионизация осуществляется при столкновении молекул исследуемого в-ва с ионами реагентного газа, в качестве к-рого м. б. индивидуальные в-ва или их смеси. Реагентный газ находится в источнике под давлением 65-130 Па, парциальное давление исследуемого в-ва 0,1-0,01 Па. При бомбардировке такой смеси электронами с энергией 70-500 эВ преим. ионизируются молекулы реагентного газа; образовавшиеся положительно заряженные ионы в результате ионно-молекулярных столкновений с неионизированными молекулами реагентного газа преобразуются в реактантные ионы, к-рые в свою очередь взаимод. с молекулами исследуемого в-ва и ионизируют их, образуя ионы МН.
Хим. ионизация с образованием положительно заряженных ионов может осуществляться также в результате переноса заряда с реактантных ионов, напр., Не+', Ar+', N2+', СО+', NO+' на молекулы исследуемого в-ва; при этом образуется мол. ион М+. Масс-спектры хим. ионизации с реагентными газами Ar и N2 напоминают спектры электронного удара. Метод хим. ионизации позволяет оценивать кислотно-основные св-ва орг. соед. в газовой фазе. Хим. ионизация с образованием отрицательно заряженных ионов осуществляется в результате взаимод. исследуемых молекул с ионами NH2, ОН, СН3О (сродство к протону соотв.1682, 816 и 778 кДж/моль). Последние образуются при захвате молекулами NH3, H2O и СН3ОН электронов с пониж. энергией (ок.6 эВ) с послед. распадом образовавшихся мол. ионов М (диссоциативный захват). Ионы ОН и СН3О образуются в значит. кол-ве при электронной бомбардировке соотв. смесей N2O с СН4 или (СН3) 3СН, Н2О и N2O с СН3ОН. Часто метод хим. ионизации более чувствительный, чем метод ионизации электронным ударом, т.к практически все имеющиеся в ионизационной камере электроны используются для ионизации. Метод позволяет анализировать пространств. и оптич. изомеры. Его важное достоинство - большой выход протонированных мол. ионов МН+ при малом выходе осколочных ионов. Полевая ионизация осуществляется в сильном электрич. поле, образующемся в пространстве между полевым анодом (острие или тонкая вольфрамовая проволока) и противоэлектродом (катодом), разность потенциалов между к-рыми 10 кВ. Молекула в таком электрич. поле теряет электрон и превращ. в положительно заряженный ион. Масс-спектры напоминают спектры электронного удара. Полевая десорбция. Труднолетучие орг. и неорг. соед. наносятся на пов-сть специально обработанного проволочного эмиттера, вблизи к-рого существует сильное электрич. поле. В результате туннельного перехода электрона молекулы к эмиттеру в-во на пов-сти проволоки ионизируется; образовавшиеся ионы десорбируются и переходят в газообразное состояние. Для облегчения десорбции проволоку подогревают, пропуская через нее электрич. ток. Применяется в анализе синтетич. полимеров и углеводородов. При фотоионизации молекулы ионизируются в результате поглощения единств. фотона, энергия к-рого должна превышать потенциал ионизации молекулы. Источники фотонов - газосветные лампы, разряды в водороде или инертных газах, синхротроны. Многофотонная ионизация газообразных в-в происходит в результате одновременного поглощения молекулой неск. фотонов. Такие процессы наблюдаются при взаимод. с в-вом достаточно интенсивного пучка лазерного излучения, энергия квантов к-рого меньше потенциала ионизации. Для этой цели используют перестраиваемые лазеры на красителях, образующие излучения с длинами волн 250-700 нм. Для ионизации большинства молекул достаточно поглощение 2-3 фотонов с энергией 1,77-4,96 эВ. Десорбционная ионизация основана на бомбардировке труднолетучего в-ва, помещенного в матрицу (глицерин, монотиоглицерин, полиэтиленгликоли, этаноламины и др. жидкости), пучками ускоренных частиц (атомы или ионы инертных газов Ar, Кr, Хе, а также ионы щелочных металлов, напр. Cs). В результате диффузионного обмена в жидкости с облучаемой пов-сти непрерывно удаляются продукты деструкции в-ва, что позволяет получать хорошо воспроизводимые масс-спектры. Применяют также метод ионизации тяжелыми продуктами деления радиоактивного 252Cf и ионами тяжелых элементов, получаемыми на ускорителях. В местах попадания таких тяжелых частиц в мишень, к-рая представляет собой пленку исследуемого в-ва на металлич. фольге, металлизир. пластике или нитроцеллюлозе, за 1011 с достигаются т-ры до 3.104 °С. Такое быстрое нагревание позволяет ионизировать тяжелые молекулы без разложения. Лазерная десорбция применяется для ионизации и испарения конденсир. в-в и осуществляется с помощью лазеров с модулированной добротностью, работающих в импульсном (длительностью до 30 нc) или непрерывном режимах. Характер масс-спектра обычно мало зависит от длины волны (265 нм - 10,6 мкм), уд. мощности (103-1010 Вт/см2) и длительности импульса лазерного излучения. Исследуемое в-во наносят на металлич. подложку и облучают фотонами с любой стороны в зависимости от конструкции прибора. Использование лазерных лучей разной степени сфокусированности позволяет проводить локальный анализ пробы в пятне диаметром 0,5 мкм - 4 мм. Возможна ионизация в-ва в искровом или тлеющем разряде. На электроды, один из к-рых изготовлен из исследуемого в-ва, подается напряжение (не более 15 кВ) в виде коротких импульсов высокой частоты. Пробой между электродами приводит к испарению материала электродов и его ионизации в образующейся плазме. Образовавшиеся положительно заряженные ионы, ускоряясь в сторону катода, к-рым служит исследуемое в-во, бомбардируют его пов-сть и распыляют образец. Распыленные частицы, проходя сквозь разряд, ионизируются. Для элементного и изотопного анализов находят применение ионные источники с ионизацией образца в индуктивно-связанной плазме Ar при атм. давлении. Поверхностная ионизация - осн. метод в изотопной М. - с. В-во наносится на пов-сть ленты из Re, W или Та, к-рая нагревается до 2000-2500 К. Если потенциал ионизации в-ва меньше работы выхода электрона из металла ленты, то часть молекул или атомов покидает ее пов-сть в ионизир. состоянии. В нек-рых случаях молекула может захватывать электрон из металла и образовывать отрицательно заряженные ионы.
Масс-анализаторы - устройства для пространств. или временного разделения ионов с разл. значениями m/z в магн. или электрич. полях или их комбинациях. Различают статич. и динамич. анализаторы. В статических ионы разделяются в постоянных или практически неизменяющихся за время их движения через анализатор магн. полях. Ионы с разл. значениями m/z движутся в таком анализаторе по разным траекториям и фокусируются либо в разных местах фотопластинки, либо последовательно на щель детектора в результате плавного изменения напряженности электрич. и магн. полей анализатора. В динамич. анализаторах разделение ионов происходит под воздействием импульсных или радиочастотных электрич. полей с периодом изменения меньшим или равным времени пролета ионов через масс-анализатор. Ионы с разл. значениями m/z, как правило, разделяются по времени пролета определенного расстояния. Давление в анализаторах должно быть достаточно низким (~105 Па), чтобы избежать рассеяния ионов на молекулах остаточных газов. Осн. характеристика масс-анализатора - его разрешающая способность, или разрешающая сила R. Она характеризует способность анализатора разделять ионы с незначительно отличающимися друг от друга массами и определяется отношением значения массы иона М к ширине его пика DМ (выраженной в атомных единицах массы) на определенном уровне высоты пика (обычно 50 или 10%): R = М/DМ. Напр., R = 10000 означает, что масс-анализатор может разделять ионы с массами 100,00 и 100,01. Hаиб. часто применяют статистические масс-анализаторы с однородным магнитным полем (одинарная фокусировка) или комбинацией электрич. и магн. полей (двойная фокусировка). В масс-анализаторах с одинарной фокусировкой (рис.4) ионный луч, сформированный в источнике ионов, выходит из щели шириной S1 в виде расходящегося ионного пучка и в магн. поле разделяется на пучки ионов с разл. значениями m/z.
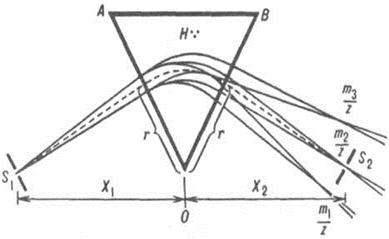
Рис.4. Схема масс-анализатора с однородным магн. полем: S1 и S2 - щели источника и детектора ионов; ОAВ - область однородного магн. поля Н, перпендикулярного плоскости рисунка; тонкие сплошные линии - границы пучков ионов с разными т/z; r - радиус центр. траектории ионов.
Под действием поля, силовые линии к-рых направлены перпендикулярно направлению движения ионного пучка, ионы двигаются по круговой траектории с радиусом r = (2Vmn/zH2) 1/2, где V - напряжение, ускоряющее ионы, mn - масса иона, z - заряд иона, H - напряженность магн. поля. Ионы с одинаковой кинетич.
энергией, но с разными массами или зарядами проходят через анализатор по разл. траекториям. Обычно развертка масс-спектра (регистрация ионов с определенными значениями m/z) осуществляется изменением Н при постоянном V. Разброс ионов, вылетающих из ионного источника, по кинетич. энергиям, а также несовершенство фокусировки по направлениям приводят к уширению ионного пучка, что сказывается на разрешающей способности. Для статич. масс-анализатора R = r/ (S1 + S2+ d), где S1 и S2 - соотв. ширина входной и выходной щелей, d - уширение пучка в плоскости выходной щели. Уменьшение размера щелей для увеличения разрешающей способности прибора трудно осуществимо технически и, кроме того, приводит к очень малым ионным токам, поэтому обычно конструируют приборы с большим радиусом траектории ионов (r = 200 - 300 мм). Разрешающая способность м. б. повышена также при использовании масс-анализаторов с двойной фокусировкой. В таких приборах ионный пучок пропускают сначала через отклоняющее электрич. поле спец. формы, в к-ром осуществляется фокусирование пучка по энергиям, а затем через магн. поле, в к-ром ионы фокусируются по направлениям (рис.5).
Рис.5. Схема масс-анализатора с двойной фокусировкой: S1 и S2 - щели источника и детектора ионов; 1 - конденсатор; 2 - магнит.
Существует более 10 типов динамич. масс-анализаторов: квадруполъный, время-пролетный, циклотронно-резонансный, магнитно-резонансный, радиочастотный, фарвитрон, омегатрон и др. Ниже рассмотрены наиб. широко применяемые масс-анализаторы. Квадрупольный масс-анализатор представляет собой квадруполъный конденсатор (Рис.6), к парам параллельных стержней к-рого приложены постоянное напряжение V и переменное высокочастотное V0cos wt (w - частота, t - время); их суммы для каждой пары равны по величине и противоположны по знаку.
Рис.6. Схема квадрупольного масс-анализатора: 1 - высокочастотный генератор; 2 - генератор постоянного напряжения; 3 - генератор развертки; 4 и 5 - источник и детектор ионов.
Ионы, вылетевшие из ионного источника, движутся в камере
анализатора вдоль оси z, параллельной продольным осям стержней, по сложным
объемным спиралевидным траекториям, совершая поперечные колебания вдоль осей x
и у. При фиксированных значениях частоты и амплитуды переменного напряжения
ионы с определенными значениями m/z проходят через квадруполъный конденсатор, у
ионов с др. значениями m/z амплитуда поперечных колебаний достигает такой
величины, что они ударяются о стержни и разряжаются на них. Развертка
масс-спектра производится путем изменения постоянного и переменного напряжении
или частоты. Для совр. квадрупольных масс-спектрометров R = 8000. Первый
квадрупольный прибор построен В. Паули и X. Штайнведелем (ФРГ, 1953). Время-пролетный
масс-анализатор представляет собой эквипотенциальное пространство, в котором
дрейфуют ионы, разделяясь по скоростям движения (рис.7). Ионы, образующиеся в
ионном источнике, очень коротким электрич. импульсом "впрыскиваются" в
виде "ионного пакета" через сетку в анализатор. В процессе движения
исходный ионный пакет расслаивается на пакеты, состоящие из ионов с одинаковыми
значениями m/z. Скорость дрейфа отслоившихся ионных пакетов и, следовательно,
время их пролета через анализатор длиной L вычисляется по ф-ле: ![]() (V
- напряжение). Совокупность таких пакетов, поступающих в детектор, образует масс-спектр.
Для совр. приборов R = 5000 - 10000. Первый прибор создан А. Камероном и Д. Эгтерсом
(США, 1948), а в СССР - Н.И. Ионовым (1956).
(V
- напряжение). Совокупность таких пакетов, поступающих в детектор, образует масс-спектр.
Для совр. приборов R = 5000 - 10000. Первый прибор создан А. Камероном и Д. Эгтерсом
(США, 1948), а в СССР - Н.И. Ионовым (1956).
Рис.7. Схема время-пролетного масс-анализатора: 1 - сетка; 2 - детектор.
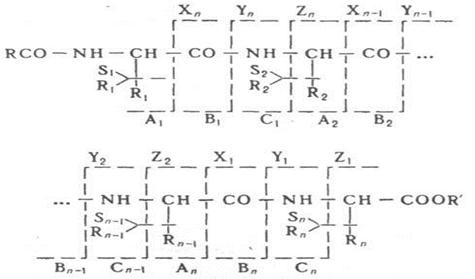
Мол. ион пептида распадается в результате разрыва связей СН-СО, СО-NH, NH-СН и СН-R с образованием осколочных ионов соотв. Аn и Хn, Вn и Yn, Сn и Zn, Sn и Rn (n - номер аминокислотного остатка в пептидной цепи), к-рые далее распадаются таким же образом. Общее кол-во пиков ионов в таком спектре может достигать неск. сотен. Кол-во фрагментов определяется строением исследуемой молекулы, запасом внутр. энергии мол. и осколочных ионов и промежутком времени между образованием иона и его детектированием. Поэтому при интерпретации масс-спектров необходимо учитывать как условия измерений (энергию ионизирующих электронов, ускоряющее напряжение, давление паров в ионном источнике, т-ру ионизац. камеры), так и конструктивные особенности прибора. При макс. стандартизации условий измерений удается получать достаточно воспроизводимые масс-спектры. Сравнение масс-спектра исследуемой системы со спектром, имеющимся в каталоге, - наиб. быстрый и простой способ структурного анализа, идентификации в-в при определении загрязнения окружающей среды, контроле продуктов питания человека и животных, изучении процессов метаболизма лек. препаратов, в криминалистике и т.д. Однако идентификация лишь на основании масс-спектра не может быть однозначной, напр. не все изомерные в-ва образуют различающиеся масс-спектры. В условиях М. - с. часть возбужденных ионов распадается после выхода из ионного источника. Такие ионы наз. метастабильными. В масс-спектрах они характеризуются уширенными пиками при нецелочисленных значениях т/z. Один из методов изучения таких ионов - спектроскопия масс и кинетич. энергий ионов. Изучение распада метастабильных ионов проводят на приборах, у к-рых магн. анализатор предшествует электрическому. Магн. анализатор настраивают таким образом, чтобы он пропустил метастабильный ион, к-рый при определенном напряжении на электрич. анализаторе проходит в детектор. Если такой ион распадается в пространстве между анализаторами, то образующиеся вторичные ионы не могут пройти через электрич. анализатор при установленном напряжении из-за недостатка энергии. Для попадания вторичных ионов в детектор изменяют напряжение электрич. анализатора. Это напряжение связано с массой вторичного иона соотношением m2 = Е2m*/Е0, где m* - метастабильный ион, m2 - вторичный ион, Е0 и Е2 - начальное и конечное напряжение электрич. анализатора. Таким образом определяются массы всех ионов, образующихся при распаде метастабильных ионов и устанавливаются тем самым схемы их фрагментации. Если в области между двумя анализаторами создать область повыш. давления (установить камеру столкновений, заполненную инертным газом), то в результате соударений ионов с молекулами газа их внутр. энергия будет увеличиваться и, следовательно, увеличится вероятность образования вторичных ионов. Такой метод, наз. тандемным, используют для структурного анализа индивидуальных компонентов сложных смесей без предварит. разделения. Наряду со структурными исследованиями М. - с. применяют для количеств. анализа орг. в-в. Количеств. анализ основан на определении интенсивностей пиков ионов с определенным значением т/z. Его проводят хромато-масс-спектрометрически (см. Хромато-масс-спектрометрия) или в системе прямого ввода. Для повышения точности определения применяют внутр. стандарты, в качестве к-рых используют меченые соед. или соед. близкие по строению к исследуемым, напр. гомологи. В последнем случае необходимо построение калибровочных кривых. Измерение содержания исследуемого в-ва проводят с учетом кол-ва добавляемого стандарта по отношению площадей пиков, соответствующих определяемому в-ву и внутр. стандарту. Погрешность метода b7%, предел определения 0,01 мкг/мл. Лучшие результаты дает применение меченых соед.; при этом отпадает необходимость в построении калибровочных кривых. Количеств. определение труднолетучих в-в проводят в системе прямого ввода, детектируя их по одному или неск. ионам, характерным для исследуемого соединения. По мере плавного повышения т-ры испарителя происходит испарение и частичное фракционирование исследуемых в-в.Т. обр., для каждого в-ва получают кривую испарения, площадь под к-рой прямо пропорциональна кол-ву соед., внесенного в масс-спектрометр. Абс. чувствительность метода, наз. методом интегрирования ионного тока, 107 г. Достоинство метода - отсутствие необходимости предварит. очистки исследуемых в-в. При исследовании соед. с электроф. группировками, изомерных орг. молекул, полимеров, азокрасителей, биологически активных в-в применяют М. - с. отрицательно заряженных ионов. Эти ионы обладают меньшим запасом внутр. энергии, чем положительно заряженные ионы, поэтому в масс-спектрах дают интенсивные пики мол. ионов и малое кол-во осколочных ионов.
Лекция 23. Блоттинг-анализ
Разработка гибридизационного метода Саузерном. Фундаментальные открытия, лежащие в основе гибридизационного метода. Перенос и идентификация макромолекул после разделения. Понятие "блоттинга". Лабораторные модификации блоттинг-метода для определения ДНК, РНК и белков. Метод in situ гибридизации, лежащий в основе гистохимических и цитохимических методов. FISH - флюоресцентная идентификация гибридизованных молекул.
Два варианта приборного и лабораторного исполнения метода полимеразной цепной реакции (ПЦР).
Современные тенденции в разработке оборудования для биохимических лабораторий - достоинства и недостатки.
© 2009 База Рефератов