
Рефераты по рекламе
Рефераты по физике
Рефераты по философии
Рефераты по финансам
Рефераты по химии
Рефераты по хозяйственному праву
Рефераты по цифровым устройствам
Рефераты по экологическому праву
Рефераты по экономико-математическому моделированию
Рефераты по экономической географии
Рефераты по экономической теории
Рефераты по этике
Рефераты по юриспруденции
Рефераты по языковедению
Рефераты по юридическим наукам
Рефераты по истории
Рефераты по компьютерным наукам
Рефераты по медицинским наукам
Рефераты по финансовым наукам
Рефераты по управленческим наукам
психология педагогика
Промышленность производство
Биология и химия
Языкознание филология
Издательское дело и полиграфия
Рефераты по краеведению и этнографии
Рефераты по религии и мифологии
Рефераты по медицине
Реферат: Прогнозирование критической температуры. Алканы и алкены
Реферат: Прогнозирование критической температуры. Алканы и алкены
Прогнозирование критической температуры
Сложность прогнозирования критической (жидкость-пар) температуры органических веществ состоит в том, что Тс изменяются нелинейно с изменением числа углеродных атомов в молекуле даже в отдельно взятой гомологической группе (рис. 5.1.). Аддитивные методы для таких свойств оказываются неэффективными, поскольку нелинейность свойства сохраняется для значительного количества соединений при переходе от низших представителей гомологических групп к высшим. Это не позволит принять некоторое постоянное значение даже для парциального вклада, характеризующего гомологическую разность, т.е. вклад на СН2 группу.
Для таких свойств широко используются аддитивно-корреляционные методы, в которых вид корреляции ответственен за изменение свойства в гомологической группе, а аддитивная составляющая свойства передает его связь со строением молекул. Рассчитывать на успех в применении этих методов возможно только в случае одинаковых соотношений типа “значение свойства - количество углеродных атомов в любой гомологической группе”. Из рис. 5.1 следует, что для критических температур это условие также не выполняется.
Приблизиться к решению проблемы удалось, используя аддитивно-корреляционные методы с дополнительной опорой на родственное с критической температурой свойство вещества. В качестве такого свойства наилучшим образом выступает нормальная температура кипения (Tb). С одной стороны, предельно близка природа этих свойств, с другой - Tb наиболее полно по сравнению с другими физико-химическими свойствами подкреплены справочными данными. Именно Tb является опорным свойством в большинстве методов прогнозирования критических температур.
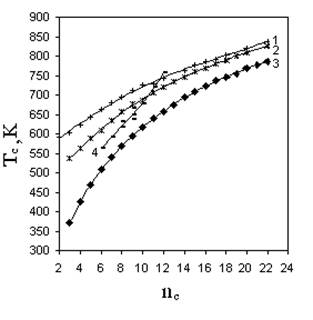
Р и с. 5.1. Зависимость критической температуры от числа углеродных атомов в молекуле:
1 - н-монокарбоновые кислоты; 2 – н-спирты;
3 – н-алканы; 4 бензол - метилбензолы
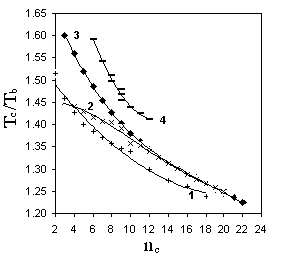
Р и с. 5.2. Зависимость Tc/Tb от числа углеродных атомов в молекуле:
1 - н-монокарбоновые кислоты;
2 – н-спирты; 3 н-алканы;
4 – бензол - метилбензолы
Иллюстрацией того, что указанный прием позволяет несколько упростить задачу прогнозирования Tс, является рис. 5.2. Однако наряду с этим из рис. 5.2 следует, что использование Tb в качестве опорного свойства не гарантирует успеха при прогнозировании Tс на основе общих универсальных корреляций для соединений любых классов. Примером тому служит совершенно иной по сравнению с соединениями прочих приведенных на рис. 5.2 классов вид корреляции для первичных спиртов С3-С10.
Метод Джобака
Сохранив основу метода Лидерсена, Джобак ввел некоторые коррективы в вид корреляции и параметризацию аддитивных составляющих свойства. В результате погрешность метода для веществ с относительно высокой молекулярной массой уменьшилась (табл. 5.1).
Корреляция, рекомендуемая для прогнозирования критических температур, требует знания нормальных температур кипения вещества и имеет вид
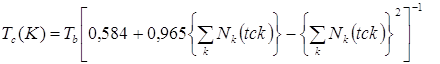 ,(5.2)
,(5.2)
где значения нормальной температуры кипения и критической температуры даны в градусах Кельвина.
Величины парциальных вкладов (tck) в критическую температуру приведены в табл. 5.3. В табл. 5.1, 5.5 приводятся результаты прогнозирования Тс методом Джобака для соединений различных классов, откуда следует, что явных преимуществ перед методом Лидерсена он все-таки не имеет. Учитывая то, что эти два метода достаточно просты в применении и в ряде случаев дают разнонаправленные отклонения, целесообразно использовать их совместно, чтобы таким образом несколько снизить погрешность в оценках критической температуры.
Следует отметить, что авторами сделана попытка увеличения точности прогноза этими методами. С этой целью был заменен вид корреляции, увеличена ее гибкость, и согласованы значения парциальных вкладов с полученной корреляцией. Однако принципиального улучшения результатов прогноза при этом не про изошло. Таким образом, возможности аддитивно-корреляционного подхода с параметризацией по атомам практически полностью использованы в методе Джобака.
Авторы сочли также целесообразным привести результаты прогнозирования Тс алканов другими методами (табл. 5.1), идеология которых не рассматривалась по следующим соображениям. Для ряда методов погрешности в оценках слишком велики уже для алканов; очевидно, что для соединений прочих классов они не станут меньше.
Метод Марреро-Пардилло дает для алканов хорошие оценки. Однако методу свойственны систематические отклонения, обусловленные избыточной жесткостью принятой авторами параболической корреляции. Аддитивная составляющая свойства формируется методом по связям аналогично тому, как это показано нами для энтальпий образования (глава 1). Использование метода по связям неизбежно повышает точность прогноза по сравнению с методами по атомам (Лидерсена и Джобака), но предъявляет серьезные требования к объему исходной информации.
Метод, основанный на индексах молекулярной связности Рандича
Поиск путей повышения точности прогноза критических температур заставил нас обратить внимание на метод, основанный на теории графов и индексах молекулярной связности Рандича [43]. Индексы Рандича наряду с другими дескрипторами более 25 лет с успехом применяются для прогнозирования различных свойств органических веществ [44-47]. Заметную роль в их практическом применении сыграли Кир и Халл [44-46]. Метод представляется нам достаточно привлекательным своей внутренней логикой. Суть метода состоит в следующем.
На основе кодовых чисел
![]() атомов в молекуле
определяются индексы молекулярной связности
атомов в молекуле
определяются индексы молекулярной связности ![]() .
Индекс молекулярной связности первого порядка равен
.
Индекс молекулярной связности первого порядка равен ![]() и отражает вклад в свойство
валентно связанных атомов. Индекс молекулярной связности второго порядка
характеризует вклад в свойство трех последовательно расположенных атомов в
молекуле и равен
и отражает вклад в свойство
валентно связанных атомов. Индекс молекулярной связности второго порядка
характеризует вклад в свойство трех последовательно расположенных атомов в
молекуле и равен ![]() . В формировании
этого и последующих индексов принимают участие не только валентно связанные
атомы, но и все те, через которые осуществляется “передача взаимодействия
атомов”. При этом степень участия “атомов-посредников” оказывается зависимой от
величины их кодовых чисел. Этот факт является, на наш взгляд, весьма сильной
стороной метода.
. В формировании
этого и последующих индексов принимают участие не только валентно связанные
атомы, но и все те, через которые осуществляется “передача взаимодействия
атомов”. При этом степень участия “атомов-посредников” оказывается зависимой от
величины их кодовых чисел. Этот факт является, на наш взгляд, весьма сильной
стороной метода.
Рандичем значения
кодовых чисел были заданы [43]. Например, для первичного, вторичного,
третичного и четвертичного атомов углерода были приняты соответственно значения
1, 2, 3 и 4. Киром и Халлом в редакции [44] значения ![]() рассчитываются на основе
количества валентных электронов и общего их числа в атоме. Таким образом, в том
и в другом случаях кодовые числа имеют постоянное значение для каждой
разновидности атомов и не зависят от прогнозируемого свойства.
рассчитываются на основе
количества валентных электронов и общего их числа в атоме. Таким образом, в том
и в другом случаях кодовые числа имеют постоянное значение для каждой
разновидности атомов и не зависят от прогнозируемого свойства.
В [25, 48] нами было
показано, что внесение некоторых изменений в методологию Кира и Халла позволяет
расширить прогностические возможности метода. Считаем принципиально важным
отказ от универсальности ![]() и
переход к кодовым числам как настраиваемым по рассматриваемому свойству
параметрам. Это позволяет более полно использовать возможности, которые
заложены в алгоритме Рандича.
и
переход к кодовым числам как настраиваемым по рассматриваемому свойству
параметрам. Это позволяет более полно использовать возможности, которые
заложены в алгоритме Рандича.
Расчет индексов молекулярной связности производится следующим образом.
1. Каждому углеродному
или гетероатому в соответствии с его типом присваивается кодовое число![]() (рис. 5.3). Значения
кодовых чисел для углеродных атомов на рисунке взяты из табл. 5.4.
(рис. 5.3). Значения
кодовых чисел для углеродных атомов на рисунке взяты из табл. 5.4.
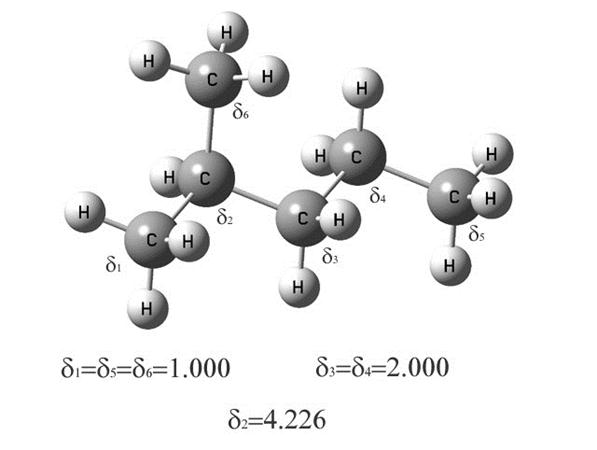
Р и с. 5.3. Кодовые числа в молекуле 2-метилпентана
2. В соответствии с
порядком i индекса молекулярной
связности ![]() в молекуле выбираются
цепочки из i+1
последовательно связанных атомов. Кодовые числа атомов, входящих в эти цепочки,
перемножаются, и индексы молекулярной связности рассчитываются как суммы этих
произведений, взятых в степени -0,5. Для приведенной молекулы 2-метилпентана
расчет индексов
в молекуле выбираются
цепочки из i+1
последовательно связанных атомов. Кодовые числа атомов, входящих в эти цепочки,
перемножаются, и индексы молекулярной связности рассчитываются как суммы этих
произведений, взятых в степени -0,5. Для приведенной молекулы 2-метилпентана
расчет индексов ![]() и
и
![]() будет выглядеть следующим
образом:
будет выглядеть следующим
образом:
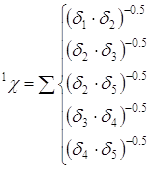
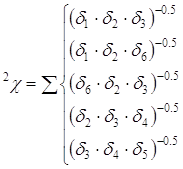
Дальнейшая работа с
индексами и прогнозируемым свойством сводится к определению вида зависимости,
адекватно описывающей экспериментальные данные. При этом в авторской редакции
[44] используется набор индексов различного порядка, которые связаны со
свойством полилинейной зависимостью. Нами использованы не индивидуальные, а
суммарные индексы молекулярной связности ![]() ,
высший порядок которых устанавливался на основе экспериментальных данных и
определял общую глубину детализации расчетного метода. При использовании в
качестве опорного свойства нормальной температуры кипения глубину детализации
метода при прогнозировании критической температуры можно ограничить 1-2.
,
высший порядок которых устанавливался на основе экспериментальных данных и
определял общую глубину детализации расчетного метода. При использовании в
качестве опорного свойства нормальной температуры кипения глубину детализации
метода при прогнозировании критической температуры можно ограничить 1-2.
Учитывая особую значимость критической температуры для прогнозирования свойств, зависящих от межмолекулярных взаимодействий, мы сочли необходимым на примере некоторых важных классов органических соединений детально изложить работу с методом и получаемые при этом результаты.
Алканы
Экспериментальные
данные по алканам (табл. 5.1) использованы следующим образом. Значения кодовых
чисел для
первичного (![]() ) и вторичного (
) и вторичного (![]() )
углеродных атомов заданы и аналогичны принятым Рандичем [43]. Значения кодовых чисел для третичных и четвертичных атомов
углерода определены одновременно с коэффициентами корреляционной зависимости
совместной обработкой всех экспериментальных данных по алканам,
приведенных в табл. 5.1.
)
углеродных атомов заданы и аналогичны принятым Рандичем [43]. Значения кодовых чисел для третичных и четвертичных атомов
углерода определены одновременно с коэффициентами корреляционной зависимости
совместной обработкой всех экспериментальных данных по алканам,
приведенных в табл. 5.1.
Анализ
получаемых при этом результатов показал, что для структур, представленных в
табл. 5.1, достаточно использовать одно значение кодового числа для третичного
углеродного атома и одно - для четвертичного. Значения их приведены в табл.
5.4. Таким
образом, для прогнозирования критических температур алканов достаточно знания
всего четырех значений кодовых чисел ![]() , два из
которых заданы, а два являются настроенными по свойству параметрами.
, два из
которых заданы, а два являются настроенными по свойству параметрами.
Таблица 5.4
Значения кодовых чисел для расчета (1-2) при прогнозировании критической температуры методом, основанным на индексах молекулярной связности
| Группа |
Кодовое число, i |
n* | Комментарий |
|
C1 |
1,000 | Задано | Углерод метильной группы во всех классах органических соединений |
|
C2 |
2,000 | Задано | Углерод метиленовой группы в насыщенных фрагментах молекул |
|
C3 |
4,226 | 32 | Третичный углеродный атом насыщенных фрагментов молекул |
|
C4 |
9,675 | 22 | Четвертичный углеродный атом насыщенных фрагментов молекул |
|
=CH2 |
1,448 | 23 | Незамещенный углерод при двойной связи в ациклической части молекул |
| =CH | 2,096 | 24 | Углерод при двойной связи в ациклической части молекул, имеющий один алкильный заместитель |
| =C | 4,398 | 6 | Углерод при двойной связи в ациклической части молекул, имеющий два алкильных заместителя |
|
Car-(H) |
3,988 | 60 | Незамещенный углерод ароматического ядра |
|
Car-(C), Car-(О) |
3,878 3,878 |
43 13 |
Замещенный углерод ароматического ядра, кроме указанного ниже случая |
| C(конденс.) | 7,896 | 5 | Узловые углеродные атомы производных нафталина |
|
Nb |
4,725 | 11 | Азот ароматического ядра пиридинов |
| OH | 0,6454 | 13 | Гидрокси-группа фенолов |
| OH | 3,540 | 17 | Гидрокси-группа первичных спиртов |
| OH | 3,540 | 10 | Гидрокси-группа вторичных спиртов |
| OH | 3,540 | 3 | Гидрокси-группа третичных спиртов |
* n-количество соединений, участвовавших в определении значений i.
При этом следует иметь в виду, что прогнозирование Тс алканов с существенно большим по сравнению с рассмотренными структурами напряжением в молекуле может потребовать расширения таблицы кодовых чисел, что несложно сделать при наличии необходимых экспериментальных данных.
Корреляционная зависимость, характеризующая связь критической температуры с нормальной температурой кипения (Tb) веществ и их индексами молекулярной связности (1-2), имеет вид
![]() ,(5.3)
,(5.3)
где ![]() - суммарный индекс
молекулярной связности второго порядка, вычисляемый по формуле
- суммарный индекс
молекулярной связности второго порядка, вычисляемый по формуле
![]() =
=![]() +
+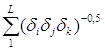 ,(5.4)
,(5.4)
а - поправка на стерическое взаимодействие атомов углерода в разветвленных углеводородах, которые разделены тремя углерод-углеродными связями. Подобный подход использован Бенсоном (гл. 1) при введении “гош-поправок” в расчет энтальпий образования органических веществ. Расчет поправки производится в едином алгоритме Рандича для всех цепочек последовательно соединенных атомов, каждая из которых включает два и более третичных и (или) четвертичных атомов углерода, по формуле
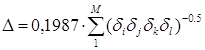 ,(5.5)
,(5.5)
что эквивалентно 0,1987×![]() для
взаимодействующих групп.
для
взаимодействующих групп.
Некоторые примеры подобных цепочек углеродных атомов приведены на рис. 5.4.
Результаты
прогнозирования ![]() алканов этим
методом, приведенные в табл. 5.1, показывают, что среднее абсолютное отклонение
расчетных значений от экспериментальных величин составляет 0,75 К, или 0,13%
отн. И это при том, что только два параметра являются настраиваемыми по
свойству, в связи с чем их оценки весьма представительны: в определении
значений
алканов этим
методом, приведенные в табл. 5.1, показывают, что среднее абсолютное отклонение
расчетных значений от экспериментальных величин составляет 0,75 К, или 0,13%
отн. И это при том, что только два параметра являются настраиваемыми по
свойству, в связи с чем их оценки весьма представительны: в определении
значений ![]() и
и ![]() участвовали 32 и 22
соединения соответственно. Максимальная погрешность расчета составляет 2,7 К,
или 0,46% отн., из 66 соединений только 5 имеют погрешность, превышающую 2 К, и
17 - 1 К.
участвовали 32 и 22
соединения соответственно. Максимальная погрешность расчета составляет 2,7 К,
или 0,46% отн., из 66 соединений только 5 имеют погрешность, превышающую 2 К, и
17 - 1 К.
Р и с. 5.4. Примеры цепочек, включаемых в расчет поправок на стерическое взаимодействие
Анализ данных табл. 5.1 показывает, что из пяти рассмотренных методов лучшие результаты для представленной выборки веществ дает метод Марреро-Пардилло. Средняя абсолютная погрешность для него составляет 1,8 К, или 0,3% отн. Методы Лидерсена, Джобака, Константину-Гани и Вильсона-Джасперсона дают соответственно 3,1, 2,4, 6,2 и 7,8 К, или 0,5, 0,4, 1,1 и 1,4% отн. При этом обращает на себя внимание тот факт, что четыре метода из пяти (Лидерсена, Джобака, Константину-Гани и Марреро-Пардилло) дают значимо смещенные оценки Tc для некоторых групп линейных алканов. В случае метода Марреро-Пардилло положительное отклонение увеличивается от 5 до 16 К при переходе от нонадекана к докозану. Источником смещенных оценок в какой-либо области гомологических групп, как уже отмечалось, является избыточная “жесткость” принятого в методе вида корреляции. Опыт показывает, что использованная нами корреляция (ур-е 5.3) обладает большей гибкостью.
Результаты прогнозирования критической температуры алкенов, ароматических углеводородов, пиридинов, фенолов, спиртов изложены ниже.
Алкены
Класс алкенов представлен 29 соединениями, 16 из которых принадлежат линейным 1-алкенам, 4 - неразветвленным геометрическим изомерам и 9 имеют разветвление в молекуле. Таким образом, база данных по Тс алкенов весьма ограниченна. Это обстоятельство необходимо учитывать при прогнозировании критических температур алкенов с более сложным строением молекулы. Для совершенствования любого метода прогнозирования необходимо пополнение базы данных, в первую очередь, сведениями для соединений с разветвленными заместителями при двойной связи и - или - углеродных атомах.
Результаты прогнозирования Тс алкенов всеми рассматриваемыми методами приведены в табл. 5.5 и являются сопоставимыми, что говорит, скорее всего, не о близких прогностических возможностях методов, а об ограниченности сведений для их оценки.
Метод, основанный на индексах молекулярной связности Рандича, реализуется с использованием всего трех дополнительных кодовых чисел для выборки веществ, приведенной в табл. 5.5. Кодовые числа, являющиеся настроенными по свойству параметрами, представлены в табл. 5.4. Количество соединений, участвующих в процессе их определения, ³ 6, что характеризует удовлетворительный уровень достоверности получаемых при прогнозе результатов.
Расчет индексов молекулярной связности выполнялся по уравнению (5.3). Аналогично алканам при расчете индексов молекулярной связности разветвленных алкенов введены поправки, учитывающие взаимодействие углеродных атомов, разделенных четырьмя связями. Поправки вычислялись в едином алгоритме Рандича для всех цепочек последовательно соединенных атомов, каждая из которых включает два и более атомов углерода, имеющих разветвление. Для таких цепочек, включающих в свой состав хотя бы один углеродный атом с разветвлением, который, к тому же, находится при двойной связи, расчет поправки производился по формуле
![]() ,(5.6)
,(5.6)
что эквивалентно 0,3334×![]() для
взаимодействующих групп.
для
взаимодействующих групп.
Таким образом, уравнение (5.6) используется для геометрических изомеров, а также для 1-алкенов с заместителями в положениях 2 и 3 или 2 и 4. Для структур с разветвлением в молекуле при и т.д. атомах углерода расчет поправки производится по уравнению (5.5), как для разветвленных насыщенных структур.
Погрешность в оценках этим методом составляет 1,3 К, или 0,2 % отн. Максимальная погрешность – 4,6 К для 1-эйкозена. Обращает на себя внимание тот факт, что алкенилбензолы не требуют самостоятельных кодовых чисел для ненасыщенных -углеродных атомов.
© 2009 База Рефератов